
Blastische plasmozytoide dendritische Zellneoplasie (BPDCN)
Die blastische plasmozytoide dendritische Zellneoplasie (BPDCN) ist eine aggressive, maligne hämatologische Erkrankung, die mit einer Häufigkeit von etwa 0,5% aller hämatologischen Neoplasien sehr selten ist. Primär treten jedoch Hautläsionen auf, weshalb eine multidisziplinäre Diagnostik essenziell ist.
Keypoints
-
Die BPDCN ist eine seltene Erkrankung. Die Diagnose muss multidisziplinär von Dermatologen, Pathologen und Hämatologen gestellt werden.
-
Essenziell ist, bei typischen Hautveränderungen an die BPDCN zu denken und die entsprechende Diagnostik einzuleiten.
-
Neben konventioneller Chemotherapie steht mit Tagraxofusp eine neue Therapieoption zu Verfügung, um Patient:innen in Remission zu bringen.
-
Nach wie vor kann nur durch eine Stammzelltransplantation ein Langzeitüberleben in Aussicht gestellt werden.
Der BPDCN liegt eine klonale Proliferation von unreifen Vorläufern plasmozytoider dendritischer Zellen zugrunde. Aufgrund der lange unklaren Abstammung dieser plasmozytoiden dendritischen Zellen variierte die Bezeichnung der Erkrankung im Lauf der Zeit (zum Beispiel blastisches NK-Zell-Lymphom, agranuläre CD4+-NK-Leukämie, Blastische-NK-Leukämie/Lymphom, agranuläre CD4+ CD56+ hämatodermische Neoplasie). Erst in der WHO-Klassifikation von 2016 wurde die BPDCN als eigenständiges Krankheitsbild unter den myeloiden Neoplasien klassifiziert. Auch in der aktuellen WHO-Klassifikation wird die BPDCN als eigenständiges Krankheitsbild unter den Neoplasien der dendritischen Zellen und Histiozyten angeführt.
Epidemiologie
Die BPDCN tritt primär bei älteren Menschen mit einer Inzidenz von etwa 0,04/ 100000 auf. Das mediane Alter der Patient:innen wird mit 61–71 Jahren angegeben. Der Prozentsatz pädiatrischer Fälle liegt bei 10–20%. Es findet sich eine männliche Prädominanz mit 3:1. Bei 20–30% der Patient:innen mit BPDCN findet sich ein vorangegangenes oder koexistentes myeloisches Malignom, zum Beispiel akute myeloische Leukämie (AML), eine myelodysplastische Neoplasie oder eine myelodysplastische/myeloproliferative Neoplasie. Die Prognose ist mit einem mittleren Überleben von rund neun Monaten ungünstig. Bei Patient:innen, die auf Therapie angesprochen haben, liegt das mediane Überleben in der Literatur bei 12–15 Monaten. Bei Kindern scheint die Prognose etwas günstiger zu sein.
Klinik und Diagnostik
Zumeist manifestiert sich die BPDCN durch Hautläsionen. So wird je nach Fallserie der Anteil der Patient:innen mit Hautbeteiligung mit 70–100% beziffert. Einzelne oder multiple erythematöse bis livide Knötchen oder ein makulopapulöser Ausschlag sind typische Veränderungen, auch hämatomartige Läsionen sind beschrieben. Häufig betroffen ist der Rumpf, bei der Hälfte der Patient:innen sind makulopapulöse Hautveränderungen an den Extremitäten und im Gesicht zu finden. Weitere Organe, die involviert sein können, sind das Knochenmark (60–90% der Fälle), das periphere Blut sowie in 40–50% der Fälle die Lymphknoten (Tab. 1).
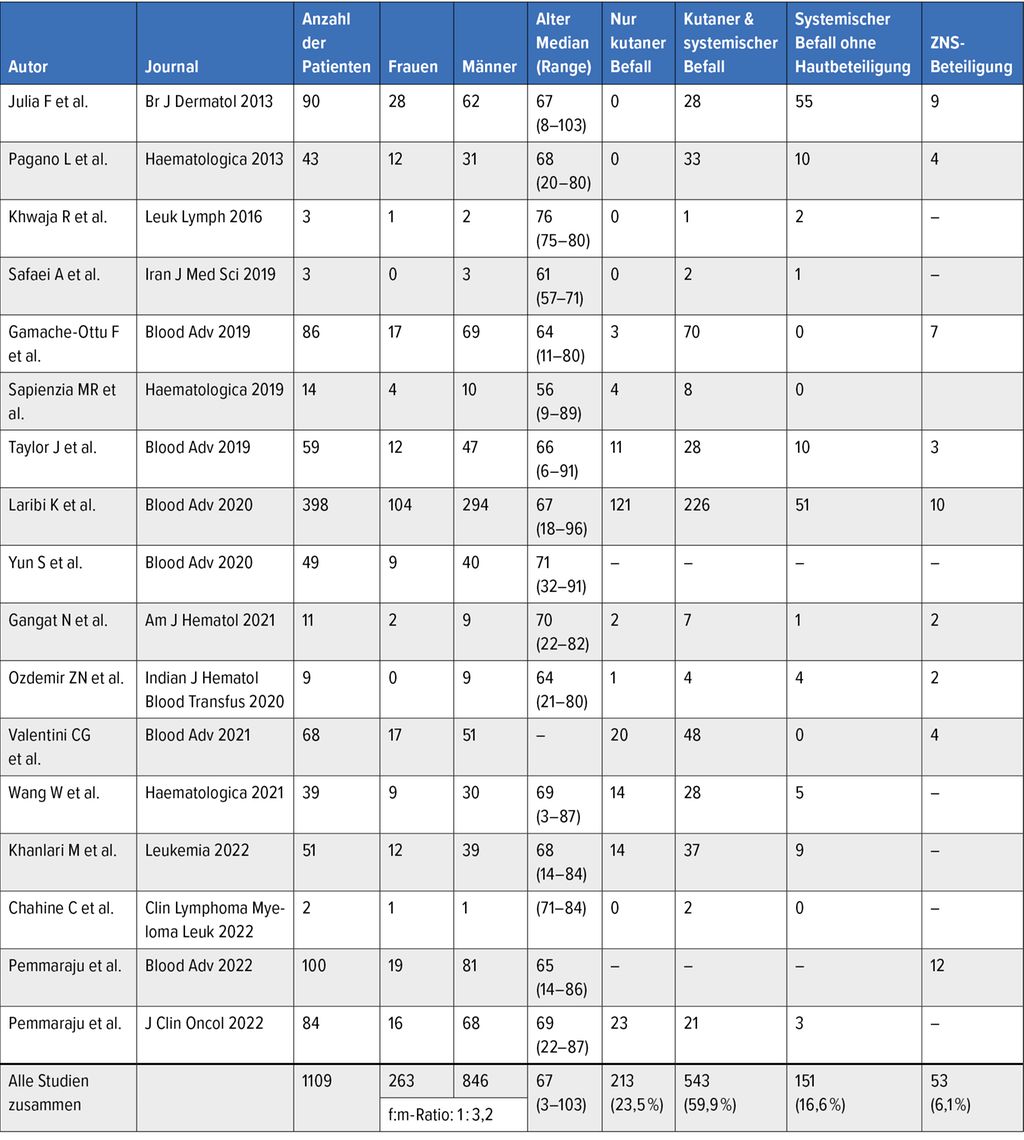
Tab. 1: Klinische Präsentation der Patienten in verschiedenen Studien
Es wird angenommen, dass die Erkrankung primär typischerweise an der Haut auftritt (90% der Patient:innen) (Tab.1). Nach einer variablen Periode relativer Stabilität folgt eine leukämische Ausbreitung mit Beteiligung multipler Organe. Diese führt zu einer raschen Verschlechterung des Allgemeinzustandes und meist in kurzer Zeit zum Tod. Selten ist die primäre Manifestation der BPDCN extrakutan zu finden.
Die Diagnose erfolgt meist durch die immunhistologische Aufarbeitung der Biopsie (Haut, Knochenmark, Lymphknoten). In dieser zeigten sich ein diffuses, monomorphes Infiltrat mittelgroßer, blastärer Zellen sowie eine hohe Anzahl von Mitosen (ca. 80%). Die Expression der Oberflächenantigene CD4, CD56, TCL1, HLA-DR, TCF4 und CD123,der α-Kette des Interleukin-3-Rezeptors, auf den blastären Zellen in der Immunhistochemie ist charakteristisch für die BPDCN. Typisch ist die Koexpression von CD123 und TCF4 auf den Zellen. Spezifische Oberflächenmarker für B-Lymphozyten (z.B. CD19), T-Lymphozyten (z.B. CD3) und myeloische Zellen (z.B. MPO, CD14) dürfen indes nicht nachweisbar sein. Die abnorme Markerexpression ist auch in der Durchflusszytometrie zu finden (Tab. 2).
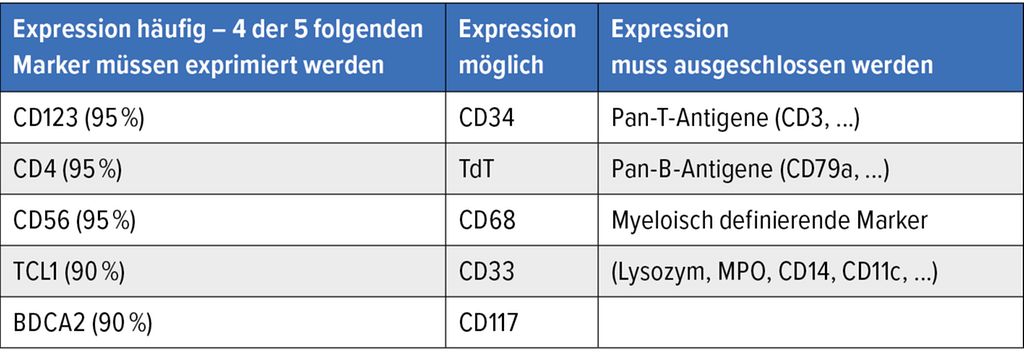
Tab. 2: Immunhistologische/durchflusszytometrische Diagnosekriterien
Wichtig im Rahmen der initialen Untersuchungen ist eine Liquordiagnostik, da sich bei etwa 10% der Patient:innen zum Zeitpunkt der Diagnose eine ZNS-Beteiligung findet. Im Verlauf der Erkrankung tritt ein Liquorbefall bei bis zu 33% der Betroffenen auf, obgleich typische Symptome oft fehlen.
Bei vielen Patient:innen bestehen auch ein komplexer Karyotyp oder chromosomale Veränderungen, die 5q (70%), 12p (62%), 13q (60%) oder 6q (52%) betreffen. Im Next-Generation-Sequencing finden sich häufig Mutationen von TET2 (21–86%), ASXL1 (28–50%), ZRSR2 (14–57%), TP53 (38%), EZH2 (14%), NRAS (14%), NOTCH1 (14%), SF3B1 (14%) und SRSF2 (14%).
Differenzialdiagnostisch ist es essenziell, BPDCN von einer AML und vice versa zu unterscheiden. Beide können in Klinik und Morphologie ähnliche Erscheinungen aufweisen, besonders, wenn bei der AML Hautinfiltrate bestehen.
Therapie
Angesichts des Verlaufs der BPDCN wurde bislang primär Chemotherapie eingesetzt. Es handelte sich dabei meist um intensive Chemotherapie, wie sie bei AML oder akuter lymphatischer Leukämie (ALL) zum Einsatz kommt, oder Standardchemotherapie wie bei Non-Hodgkin-Lymphomen (NHL) (Tab. 3). Mit allen Protokollen konnten bei einem Teil der Patient:innen Remissionen erreicht werden. Angesichts der Seltenheit der Erkrankung sind bisher keine randomisierten Studien publiziert. Vielmehr stützt sich unsere Beurteilung zur Effektivität unterschiedlicher Therapieschemata auf retrospektive Daten. Generell konnten mit intensiven Induktionstherapien im Vergleich zu Standardtherapien (NHL-Protokolle) höhere Remissionsraten erreicht werden. Außerdem scheinen ALL-Protokolle den AML-Protokollen in Bezug auf die Ansprechraten überlegen zu sein.
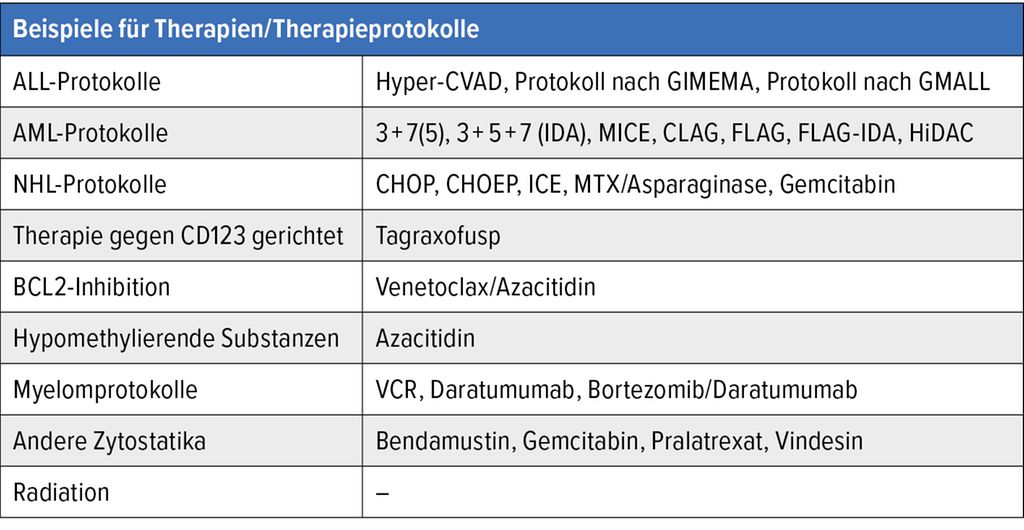
Tab. 3: Beispiele für Therapien/Therapieprotokolle, die bei BPDCN zur Anwendung kamen und kommen
Trotz des moderaten bis guten Ansprechens bedingt eine hohe Rezidivrate ein überschaubares Langzeitüberleben, wie Laribe K et al. 2022 in einer retrospektiven Datenanalyse von 398 Patient:innen aus 75 Zentren zeigte. Das Überleben der Patient:innen betrug mit ALL/AML-Protokollen 18 Monate und war nur unwesentlich länger als unter NHL-Protokollen (14 Monate).
Die einzige Therapieoption, die zu einer signifikanten Verlängerung des Überlebens führt, ist die Stammzelltransplantation. Dies konnte für die allogene wie auch für die autologe Stammzelltransplantation gezeigt werden. Dabei ist die autologe der allogenen Transplantation unterlegen. Entsprechend ist eine Stammzelltransplantation, wenn möglich, die Therapie der Wahl, wobei das vorhergehende Erreichen einer kompletten Remission für das Langzeitüberleben essenziell ist.
Vor Kurzem wurde Tagraxofusp zur Therapie der BPDCN zugelassen. Dabei handelt es sich um ein Fusionsprotein aus rekombinantem IL-3 und einem verkürzten Diphtherietoxin, das sich die Expression von CD123 auf den Zellen der BPDCN zunutze macht. Die Effektivität der Therapie konnte 2019 in einer Open-Label-Studie gezeigt werden. Von 29 Patient:innen, die nach Dosisfindung mit 12μg/kg Körpergewicht Tagraxofusp behandelt worden waren, zeigten 72% ein vollständiges oder „klinisch vollständiges“ Ansprechen. In den 2022 publizierten Langzeitdaten von 65 Patient:innen lag das vollständige oder „klinisch vollständige“ Ansprechen bei 57% und das Gesamtansprechen (inklusive partieller Response) bei 75%. Bei 45% konnte nach Erreichen einer Remission eine Stammzelltransplantation durchgeführt werden. Zu erwähnen ist, dass ein ECOG-Score <2 und eine adäquate Organfunktion Voraussetzung für den Einschluss in die Studie waren. Vor allem im ersten Therapiezyklus traten Nebenwirkungen wie Anstieg der Transaminasen, Hypoalbuminämie, periphere Ödeme, Thrombozytopenie und Capillary-Leak-Syndrom auf. Bei Letzterem war ein Abfall des Albumins am ersten Behandlungstag zu beobachten. Entsprechend empfehlen die Leitlinien, bei Auftreten eines Albuminabfalls die weiteren Tagraxofusp-Gaben zu pausieren und Albumin zu substituieren.
Auch eine Reihe von anderen neuen Therapien wurde bei der BPDCN bereits erfolgreich eingesetzt, wobei bis dato diesbezüglich nur Fallserien oder Fallberichte publiziert wurden. Prominente Beispiele wären Venetoclax/Azacitidin, Azacitidin-Monotherapie oder Bortezomib/Daratumumab.
Bei Patient:innen mit ZNS-Beteiligung ist eine entsprechende intrathekale Therapie wie bei ALL erforderlich.
Zusammenfassung
Die BPDCN ist eine seltene Erkrankung. Die Diagnose muss multidisziplinär von Dermatologen, Pathologen und Hämatologen gestellt werden. Essenziell ist, bei typischen Hautveränderungen an die Erkrankung zu denken und die entsprechende Diagnostik einzuleiten. Neben konventioneller Chemotherapie steht mit Tagraxofusp eine neue Therapieoption zu Verfügung, um Patienten in Remission zu bringen. Nach wie vor kann nur durch eine Stammzelltransplantation ein Langzeitüberleben in Aussicht gestellt werden.
Literatur:
bei den Verfasser:innen