Polycythaemia vera – Aderlass ist out?
In der Therapie der Polycythaemia vera (PV) ist der Aderlass noch immer ein Mittel der Erstlinientherapie, führt jedoch langfristig zu Eisenmangel. Neue Therapien wie die JAK1/2-Inhibition mit Ruxolitinib oder Ropeginterferon alfa-2b sind ein bedeutender Fortschritt in der langfristigen PV-Behandlung, da sie kontinuierlich die Allellast senken und das ereignisfreie Überleben verlängern.
Keypoints
-
Alleinige Aderlasstherapie in Kombination mit ASS adressiert nicht die Krankheitsprogression zu Myelofibrose oder AML.
-
Die Rolle des Aderlasses liegt in der schnellen initialen Hämatokritkontrolle zur Vermeidung von akuten thromboembolischen Ereignissen.
-
Eine langfristige Aderlasstherapie führt zu Eisenmangelsymptomen mit Einschränkung der Lebensqualität.
-
Das krankheitsmodifizierende Potenzial von Interferon alfa konnte sowohl für Hochrisiko- als auch für Niedrigrisiko-Patient:innen gezeigt werden.
Seit der Erstbeschreibung der Polycythaemia vera (PV) 1892 kommt traditionell der Aderlass als therapeutische Option zum Einsatz. Auch heute noch wird bei der überwiegenden Mehrheit der Patient:innen mit neu diagnostizierter PV der Aderlass als relativ schnell wirksame Erstlinientherapie angewandt, um Akutkomplikationen wie Myokardinfarkt oder Schlaganfall zu verhindern. Durch wiederholte Aderlässe kann bei einem Teil der Patient:innen bei alleiniger Anwendung der Hämatokrit unter dem definierten Zielwert von 45% gehalten werden, ohne jedoch in die komplexe Pathophysiologie der Erkrankung einzugreifen. Ein Nachteil der langfristigen Aderlasstherapie sind die sich einstellenden Symptome des Eisenmangels.
Molekulares Ansprechen und längeres EFS unter JAK-Inhibition
Die 2005 bei myeloproliferativen Neoplasien (MPN) erstbeschriebene JAK2-Mutation V617F ist bei über 95% der PV-Patient:innen nachweisbar und führt zu einer konstitutionellen Aktivierung des JAK-STAT-Signalweges und zu hämatopoetischer Hyperproliferation.1 Ziele der Therapie sind neben dem Vermeiden thromboembolischer Komplikationen die Verbesserung der Lebensqualität und vor allem eine Modifikation des Krankheitsverlaufes.
Der JAK1/2-Inhibitor Ruxolitinib wurde bei Polycythaemia vera randomisiert geprüft gegenüber BAT („best available therapy“) in der Phase-II-Studie MAJIC-PV2 und der Phase-IIIb-Studie RESPONSE-2.3,19 Ruxolitinib führt zu einer verbesserten und anhaltenden hämatologischen Kontrolle (CR nach ELN-Kriterien), zu einer effektiven Symptomkontrolle und Verbesserung der Lebensqualität. Im Vergleich zu BAT benötigten nur etwa die Hälfte der Patient:innen im Ruxolitinib-Arm zusätzlich mindestens einen Aderlass zur Hämatokrit-Kontrolle (Ziel ≤45%).2
Anlässlich des 5-Jahres-Follow-up der MAJIC-PV-Studie konnte 2023 gezeigt werden, dass unter Ruxolitinib das ereignisfreie Überleben (EFS) signifikant verlängert wird. Zudem wird ein molekulares Ansprechen erreicht: Abnahme der JAK2-Allellast („variant allele frequency“, VAF) im peripheren Blut. Die mediane Zeit bis zum molekularen Ansprechen unter Ruxolitinib lag bei 36 Monaten versus nicht erreicht im BAT-Arm.2
Ropeginterferon alfa-2b senkt Allellast kontinuierlich
Ropeginterferon alfa-2b hat seit 2019 die EU-Zulassung in der Erstlinie der PV-Therapie. Interferon (IFN) wirkt ebenfalls über den JAK-STAT-Signalweg und führt bei Langzeitbehandlung zu einer kontinuierlichen Abnahme des JAK2-V617F-Allelburden. In den Studien PROUD-PV und CONTINUATION-PV erfolgte der Vergleich von IFN mit Hydroxyurea (HU) im ersten Therapiejahr und mit BAT (davon 88% persistierend HU) ab Jahr 2. Das Ansprechen auf Ropeginterferon alfa-2b verbessert sich kontinuierlich mit der Dauer der Behandlung bei guter Verträglichkeit der subkutanen Gaben im zweiwöchentlichen Abstand.5 In der finalen Auswertung der sechsjährigen Verlaufsbeobachtung war nicht nur der Anteil der Patient:innen mit anhaltender kompletter hämatologischer Remission (CHR) im Interferonarm signifikant höher (72,6% vs. 52,6%, p=0,004), sondern auch das EFS (Thromboembolien, Krankheitsprogression oder Tod) verlängert und das molekulare Ansprechen (69,1% vs. 21,6%, p<0,0001) gegenüber BAT verbessert.5,10 Etwa die Hälfte der Patient:innen (50 von 92) zeigte eine Reduktion der Allellast auf <10% und 14 von 92 Patient:innen eine Allellast von <1% nach 72 Monaten der Therapie mit Ropeginterferon alfa-2b.10
Die Abnahme des JAK2-V617F-Allelburden dient als Surrogatmarker für die Krankheitsmodifikation, obwohl bislang prospektive Studien zur Frage fehlen, inwiefern eine Senkung der JAK2-Allellast zu einer Prävention der Progression zu Myelofibrose oder MDS/AML führt. Bezüglich der Krankheitsprogression ist zu beachten, dass bei >50% der PV-Patient:innen bereits bei Diagnosestellung subklonale Nondriver-Läsionen nachweisbar sind, wie TET2 (keine prognostische Bedeutung), ASXL1 und LNK. Im Verlauf können weitere MDS-/AML-typische Mutationen hinzukommen, wie SRSF2-, RUNX1-, TP53- oder IDH-Mutationen. Diese gehen neben Alter, erhöhter Leukozytenzahl und abnormalem Karyotyp in den „mutation-enhanced international prognostic score“ (MIPPS-PV) ein. Die resultierenden Risikogruppen „low“, „intermediate-1“, „intermediate-2“ und „high“ trennen die Überlebenswahrscheinlichkeit in ein Spektrum von median 5,4 Jahren für die Hochrisikogruppe und median 25,3 Jahren für die Niedrigrisikogruppe auf.20
Die individuell optimale Therapiefinden
Die Herausforderung besteht in einer maßgeschneiderten Therapie. Die oft belastenden Symptome bei PV sind Pruritus, konstitutionelle Symptome wie ausgeprägte Fatigue, Mikrozirkulationsstörungen wie Erythromelalgie und das Auftreten thromboembolischer Ereignisse trotz der Therapie mit ASS und Hämatokrit-Senkung unter 45% gemäß den Leitlinienempfehlungen. Die klassische Einteilung in Niedrigrisiko-PV und Hochrisiko-PV bezieht sich auf die Wahrscheinlichkeit für Thromboembolien und umfasst lediglich zwei Faktoren: Alter ≥60Jahre und das Vorhandensein thromboembolischer Ereignisse in der Vorgeschichte. Weitere Faktoren für Thrombose und für Krankheitsprogression sind Leukozytose und eine hohe JAK2-V617F-Allellast, sie gehen aber nicht in die Risikoklassifikation ein.16,17
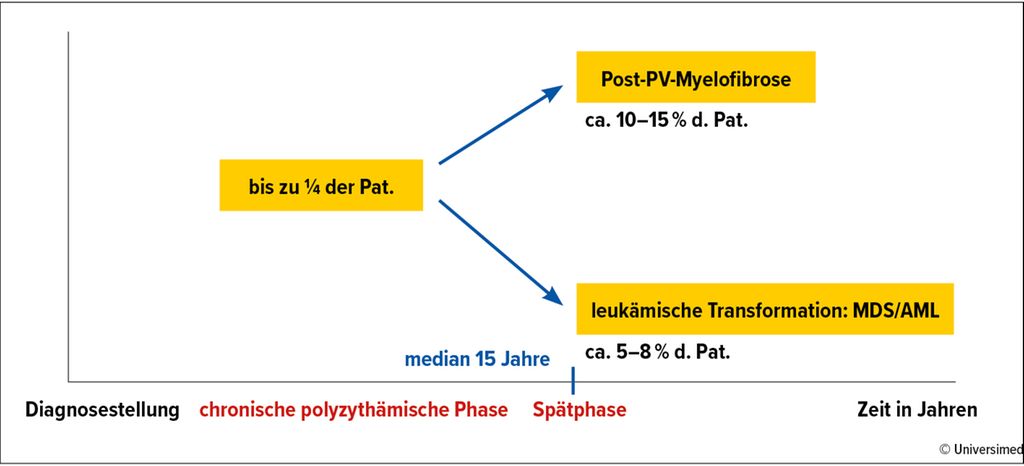
Abb. 1: Progressionsrisiko bei PV
Durch alleinige Aderlässe in Kombination mit ASS oder die Hinzunahme von Hydroxyurea ist weder eine befriedigende Kontrolle der Symptomlast noch die Vermeidung thromboembolischer Komplikationen im Langzeitverlauf zu erreichen. Daher rückt der frühe Einsatz krankheitsmodifizierender Therapien wie Ropeginterferon oder Ruxolitinib in den Fokus.
Ruxolitinib ist seit 2014 in der Zweitlinie zugelassen bei Resistenz oder Intoleranz gegenüber Hydroxycarbamid.14 Der Wirkstoff ist besonders geeignet zur Symptomkontrolle und zur Reduktion der Milzgröße.
Ropeginterferon ist zugelassen als Monotherapie bei PV ohne symptomatische Splenomegalie unabhängig von der Vortherapie oder vom Vorhandensein von Risikofaktoren für thromboembolische Ereignisse.15 Der Einsatz von Ropeginterferon wird sowohl bei geeigneten Hochrisiko-Patient:innen als auch bei Niedrigrisiko-Patient:innen empfohlen.11–13
Innerhalb des spanischen Polycythaemia-vera-Registers wurden über zehn Jahre (2011–2021) 453 Niedrigrisiko-Patient:innen beobachtet, die initial ausschließlich mit Aderlass und ASS behandelt wurden. Eine anhaltende Hämatokrit-Kontrolle war nur bei einem Drittel der Patient:innen zu erreichen und eine befriedigende Symptomkontrolle im Verlauf wurde nicht erzielt. Thrombotische Ereignisse traten mit einer Häufigkeit von 0,8% pro Jahr auf (erwartete Wahrscheinlichkeit von 8,5% in zehn Jahren). Es zeigte sich ein Trend zu einem höheren Risiko für venöse Thrombosen bei hohem JAK2-Allelburden.18 Auf der Basis der Low-PV-Studie, einer randomisierten Phase-II-Studie, die Ropeginterferon alfa-2b versus Phlebotomie vergleicht, wurde Ropeginterferon auch zur Therapie der Niedrigrisiko-PV zugelassen mit dem Ziel der frühen Krankheitsmodifikation.6,21
Rolle des Aderlasses hat sich gewandelt
Ein Problem der alleinigen Aderlass-Therapie in Kombination mit ASS ist auch der schwankende Hämatokritwert in der Längsschnittanalyse, da der Aderlass immer nur punktuell den Erythrozyten-Overload „abschneidet“. Damit ist kein durchgängiges hämatologisches Ansprechen zu erzielen. In einer retrospektiven „Real-world“-Analyse von 28306 PV-Patient:innen (2011–2019) zeigten sowohl die Patient:innen der Niedrig- als auch der Hochrisiko-Gruppe unter Aderlass-Monotherapie eine suboptimale Hämatokrit-Kontrolle (definiert als Hk >50% manchmal oder immer). Dies betraf 54% der Patient:innen in der Hochrisiko-Gruppe und 64% in der Niedrigrisiko-Gruppe.22 Neben diesem unbefriedigenden Management in Bezug auf das Verhindern vaskulärer Komplikationen und Reduzieren krankheitsspezifischer Symptome wird in der Langzeitperspektive das Vermeiden der Krankheitsprogression nicht adressiert.
Damit hat sich die Rolle des Aderlasses grundlegend gewandelt und liegt in der initialen Hämatokrit-Senkung, um akute vaskuläre Komplikationen zu verhindern. Zur Langzeitkontrolle der Erkrankung ist ein früher Beginn einer zytoreduktiven Therapie indiziert mit Präferenz von Ropeginterferon alfa-2b.11,12
Therapieziele bei Polycythaemia vera
Vermeidung thromboembolischer Komplikationen im Langzeitverlauf
Senkung des Risikos der Krankheitsprogression zu Myelofibrose oder akuter Leukämie
Verbesserung der Lebensqualität durch Reduktion der Symptomlast
Vermeidung von Aderlässen, um Eisenmangelsymptome zu vermeiden
Literatur:
1 Baxter EJ et al.: Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365(9464): 1054-61 2 Harrison CN et al.: Ruxolitinib versus best available therapy for polycythemia vera intolerant or resistant to hydroxycarbamide in a randomized trial. JCO 2023; 41(19): 3534-44 3 Kiladjian JJ et al.: Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol 2020; 7(3): e226-37 4 Barosi G et al.: Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 2013; 121(23): 4778-81 5 Gisslinger H et al.: Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol 2020; 7(3): e196-208 6 Barbui T et al.: Ropeginterferon alfa-2b versus phlebotomy in low-risk patients with polycythaemia vera (Low-PV study): a multicentre, randomised phase 2 trial. Lancet Haematol 2021; 8(3): e175-84 7 Abu-Zeinah G et al.: Interferon-alpha for treating polycythemia vera yields improved myelofibrosis-free and overall survival. Leukemia 2021; 35(9): 2592-601 8 Sørensen AL et al.: Ruxolitinib and interferon-α2 combination therapy for patients with polycythemia vera or myelofibrosis: a phase II study. Haematologica 2020; 105(9): 2262-72 9 Kiladjian JJ et al.: Long-term outcomes of polycythemia vera patients treated with ropeginterferon alfa-2b. Leukemia 2022; 36(5): 1408-11 10 Gisslinger H et al.: Event free survival in patients with polycythemia vera treated with ropeinterferon alfa-2b versus best available treatment. Leukemia 2023; 37(10): 2129-32 11 Lengfelder E et al.: Onkopedia Leitlinie Polycythaemia vera (PV) Stand September 2023 ( https://www.onkopedia.com/de/onkopedia/guidelines/polycythaemia-vera-pv/@@guideline/html/index.html ) 12 NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Myeloproliferative Neoplasms. Version 1.2024 ( https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf ) 13 Marchetti M et al.: Appropriate management of polycythaemia vera with cytoreductive drug therapy: European LeukemiaNet 2021 recommendations. Lancet Haematol 2022; 9(4): e301-11 14 Jakavi Fachinformation, Stand April 2022 15 Besremi Fachinformation, Stand Februar 2019 16 Vannucchi AM et al.: Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 2007; 21(9): 1952-9 17 Carobbio A et al.: JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol 2009; 37(9): 1016-21 18 Triguero A et al.: Low-risk polycythemia vera treated with phlebotomies: clinical characteristics, hematologic control and complications in 453 patients from the Spanish registry of polycythemia vera. Ann Hematol 2022; 101(10): 2231-9 19 Passamonti F et al.: Ruxolitinib versus best available therapy in inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): 5-year follow up of a randomised, phase 3b study. Lancet Haematol 2022;9(7): e480-92 20 Tefferi A et al.: Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol 2020; 189(2): 291-302 21 Barbui T et al.: Ropeginterferon versus standard therapy for low-risk patients with polycythemia vera. NEJM Evid 2023; 2(6): EVIDoa2200335 22 Verstovsek S et al.: Real-world treatments and thrombotic events in polycythemia vera patients in the USA. Ann Hematol 2023; 102(3): 571-81
Das könnte Sie auch interessieren:

Erfolg zielgerichteter und zeitlich begrenzter Therapien
Auf der Frühjahrstagung der OeGHO widmete sich eine spezielle Session zukunftsweisenden Entwicklungen in der Grundlagen- und klinischen Forschung auf dem Gebiet der Hämatologie. Im Fokus ...

Tafasitamab wirksam bei Patient:innen mit multiplen Risikofaktoren
Mit einer Post-hoc-Analyse der Phase-III-Studie inMIND wurden die Vorteile für den Einsatz von zusätzlichem Tafasitamab zu Lenalidomid plus Rituximab beim follikulären Lymphom für bisher ...

Erworbene Hämophilie A
Die erworbene Hämophilie A zeigt sich mit einer Inzidenz von 1,5–1,8pro Million pro Jahr. Es kommt zu spontanen, teilweise schweren und lebensbedrohlichen Blutungsereignissen aufgrund ...