ATTR-CM – von einer unheilbaren zu einer behandelbaren Erkrankung
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Für die Behandlung der Transthyretin-Amyloid-Kardiomyopathie (ATTR-CM) gab es bis vor Kurzem noch keine zugelassene spezifische Therapie. Neben den vielversprechenden Ergebnissen der Zulassungsstudie zum Wirkstoff Tafamidis konnte im Rahmen einer explorativen Analyse gezeigt werden, dass sich eine Behandlung positiv auf die NT-proBNP-Konzentration im Serum sowie auf die Leistungsfähigkeit von Patienten mit ATTR-CM auswirken kann. Der folgende Artikel gibt Einblick in diese hoffnungsvolle neue Therapieoption.
Keypoints
Die Transthyretin-Amyloid-Kardiomyopathie (ATTR-CM) ist durch zunehmende Ablagerungen von Amyloidfibrillen im Herzmuskel bedingt.
Neue Therapieansätze durch Stabilisierung des Transthyretin-Proteins sind in der Lage, die Progression der Erkrankung zu verlangsamen.
Standen noch vor wenigen Jahren keine spezifischen Wirkstoffe zur Verfügung, so wandelte sich die ATTR-CM durch Zulassung neuer Therapieoptionen von einer unheilbaren zu einer behandelbaren Erkrankung.
Pathophysiologie
Als Amyloide bezeichnet man fehlgefaltete Proteine mit charakteristischer Cross-β-Struktur, welche sich durch enzymatische Umwandlungsprozesse als unlösliche Komplexe in Form von Amyloidfibrillen in diversen Organen ablagern und so zu deren Dysfunktion im Sinne der Erkrankung Amyloidose führen.1 Zu den häufigsten Formen der kardialen Amyloidosen zählt die Transthyretin-Amyloid-Kardiomyopathie (ATTR-CM), deren Pathogenese von Transthyretin (TTR), einem hepatisch synthetisierten Transportprotein für Vitamin A und Schilddrüsenhormone, dominiert wird. Im Rahmen der Erkrankung lässt sich eine nicht erblich bedingte, altersassoziierte Wildtyp-Form (wtATTR) von einer autosomal-dominant vererbbaren –hereditären – Form (hATTR) abgrenzen.2 Während die Pathogenese der wtATTR noch nicht vollständig geklärt ist,3 kommt es bei der hATTR bedingt durch Punktmutationen im TTR-Gen zur Destabilisierung des TTR-Tetramers und zur Dissoziation in seine Monomere. Diese diffundieren in das umliegende Gewebe und bilden durch enzymatische Umwandlungsvorgänge unlösliche Komplexe in Form von Amyloidfibrillen.2
Klinische Präsentation
Die Ablagerung von Amyloidfibrillen im Herzmuskel führt zu einer Verdickung des Myokards, welche mit einer erhöhten Steifigkeit mit begleitender diastolischer und systolischer Funktionseinschränkung einhergeht.3 Klinisch präsentieren sich betroffene Patienten oftmals mit Symptomen der globalen Herzinsuffizienz, welche meist mit dem Auftreten von Belastungsdyspnoe, Pleuraergüssen und Beinödemen sowie Hepatomegalie, Aszites und einem erhöhten Jugularvenendruck assoziiert ist.4 Des Weiteren können Amyloidablagerungen in den Herzkranzgefäßen zu Angina-pectoris-Symptomen führen.5 Einlagerungen im Reizleitungssystem können einerseits Schwindel und Synkopen begünstigen, sich andererseits aber auch als lebensbedrohliche ventrikuläre Arrhythmien manifestieren.6 Ein zusätzlicher Befall des autonomen Nervensystems kann zu autonomen Störungen wie orthostatischer Hypotonie und gastrointestinaler Dysmotilität führen, während Ablagerungen im peripheren Nervensystem durch Polyneuropathien und/oder das Auftreteneines Karpaltunnelsyndroms gekennzeichnet sind.7
Diagnose
Die klinische Präsentation des Patienten wie auch laborchemische Veränderungen im Sinne einer Erhöhung der Biomarker NT-proBNP und Troponin T sowie Veränderungen im EKG (z.B. QRS-Niedervoltage, Vorhofflimmern) können schon erste Hinweise auf das Vorliegen einer ATTR-CM liefern.5 Die weitere Abklärung wird von bildgebenden Verfahren dominiert, wobei die Echokardiografie aufgrund ihrer breiten Verfügbarkeit eine zentrale Rolle spielt. Zu den sogenannten „red flags“, die auf eine ATTR-CM hinweisenkönnen, zählt die biventrikuläre Verdickung mit einem hypertrophierten interatrialen Septum und vergrößerten Vorhöfen. Zusätzlich finden sich auch häufig Perikard- und/oder Pleuraergüsse. Darüber hinaus erlaubt der Global Longitudinal Strain (GLS) eine zuverlässige Beurteilung der linksventrikulären Funktion, wobei ein vor allem basal reduzierter GLS („apical sparing“) eine diagnostische Schlüsselrolle spielt (Abb. 1 A, B).4Außerdembietet die kardiale Magnetresonanztomografie die Möglichkeit, mithilfe von „LateEnhancement“-Aufnahmen und T1-Mapping-Sequenzen Amyloidablagerungen im Myokard zu quantifizieren (Abb.1, C–E).8 Die wichtigste nichtinvasive bildgebende Untersuchungsmethode zur Diagnose einer ATTR-CM ist jedoch die Knochenszintigrafie mit radioaktiv markierten Tracern (z.B. 99mTc-DPD; Abb.1, F).9
Neben den bildgebenden Verfahren bildet die Serum- und Urinanalyse eine weitere diagnostische Säule. Dabei können das Vorliegen einer pathologischen Kappa-/Lambda-Ratio oder das Vorliegen von Paraprotein in der Serum- und/oder Harndiagnostik auf eine AL-Amyloidose hinweisen, welche differenzialdiagnostisch jedenfalls in Betracht gezogen werden sollte.10 Aufgrund des breiten Spektrums an diagnostischen Möglichkeiten ist heutzutage eine korrekte Diagnosefindung in den meisten Fällen auch ohne Endomyokardbiopsie möglich. Bei unklaren Befundkonstellationen oder speziellen Fragestellungen gilt die Herzmuskelbiopsie jedoch nach wie vor als Goldstandard in der Amyloidosediagnostik.10
Therapeutischer Ansatz
Da die Dissoziation des TTR-Tetramers in Monomere einer der wichtigsten pathophysiologischen Schritte bei der ATTR-CM ist, ist die Stabilisierung des Tetramers ein valider therapeutischer Ansatz. Tafamidis fungiert mittels kleiner Moleküle als Stabilisator und wirkt somit in weiterer Folge der Bildung von Amyloidfibrillen und ihren Ablagerungen im Herzmuskel entgegen. Dies wiederum trägt womöglich zu einer Verlangsamung der Progression der Erkrankung bei.
Therapeutischer Ausblick
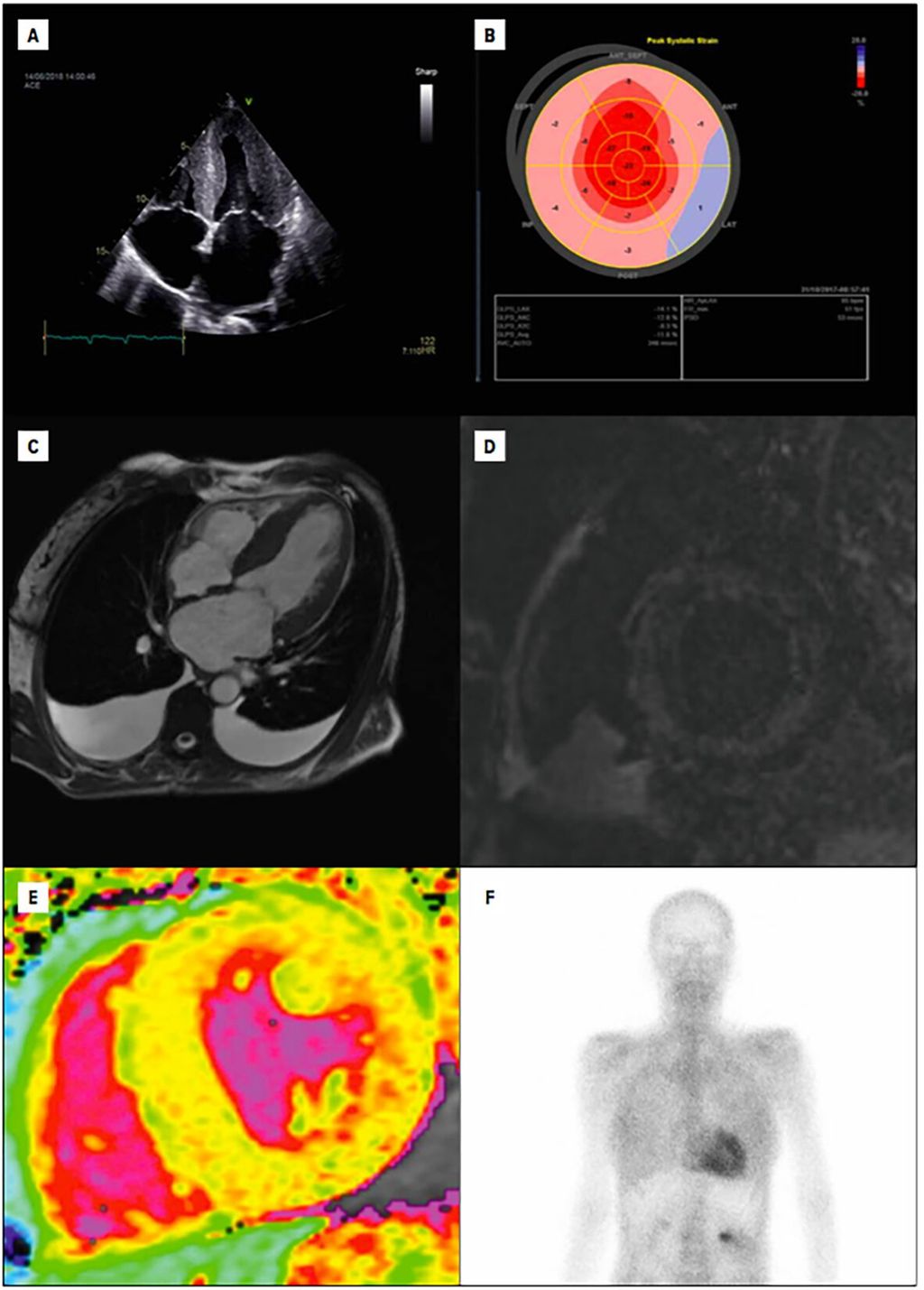
Abb. 1: Bildgebende Verfahren zur Diagnostik einer Transthyretin-Amyloid-Kardiomyopathie (ATTR-CM). Bilder A und B zeigen echokardiografische Charakteristika der ATTR-CM. A zeigt einen 4-Kammer-Blick mit dem Vollbild einer ATTR-CM: Linksventrikelhypertrophie und vergrößerte Vorhöfe. In B ist eine Strain-Analyse des linken Ventrikels mit dem für die ATTR-CM typischen „apical sparing“ zu sehen.
Die Bilder C–E zeigen magnetresonanztomografische Charakteristika der ATTR-CM. In C ist ein 4-Kammer-Blick mit dem Vollbild einer ATTR-CM zu sehen. D zeigt eine „Late gadolinium enhancement“-Aufnahme in der kurzen Achse mit diffus-fleckiger myokardialer Kontrastmittelaufnahme. E zeigt eine „native T1 map“ in der kurzen Achse mit stark erhöhten myokardialen T1-Zeiten (hellgelb bis rötlich), welche typisch für eine ATTR-CM sind. In F ist eine Knochenszintigrafie mit ausgeprägter myokardialer Traceranreicherung zu sehen.
Im Rahmen einer placebokontrollierten, internationalen, multizentrischen Phase-III-Studie mit 440 ATTR-CM-Patienten konnten signifikante Vorteile hinsichtlich dekompensationsbedingter Hospitalisierungen (52,3% vs. 60,5%) als auch in Bezug auf die Gesamtmortalität (29,5% vs. 42,9%) durch eine Behandlung mit Tafamidis aufgezeigt werden.11 Eine explorative Analyse der klinischen Abteilung für Kardiologie an der Medizinischen Universität Wien beleuchtete die Auswirkungen von Tafamidis im Vergleich zu einer Kohorte ausgewählter ATTR-CM-Patienten ohne spezifische Therapie. Im Rahmen dieser Studie wurden 38 Patienten mit diagnostizierter ATTR-CM für einen Zeitraum von 6 Monaten mit Tafamidis 20mg bzw. 61mg behandelt. Dabei konnte im Vergleich zur unbehandelten Kontrollkohorte eine signifikante Reduktion der Serumkonzentration von NT-proBNP festgestellt werden. Dies spiegelte sich auch in einer gesteigerten Leistungsfähigkeit durch ein besseres Abschneiden während des 6-Minuten-Gehtests im Kohortenvergleich wider. Des Weiteren zeigten sich bei den bildgebenden Parametern (Echokardiografie, kardiale Magnetresonanzomografie) leichte kardiostrukturelle Verbesserungen im Vergleich zur unbehandelten Kontrollkohorte. Aufgrund der vielversprechenden Ergebnisse der Phase-III-Zulassungsstudie ATTR-ACT11 liegt seit dem zweiten Quartal 2020 eine europäische Zulassung von Tafamidis zur Therapie der Wildtyp- oder hereditären ATTR-CM vor, wodurch sich eine bisher unheilbare zu einer behandelbaren Erkrankung gewandelt hat.
Literatur:
1 Meinhardt J et al.: Struktur von Amyloidfibrillen. Pathologe 2009; 30(3): 175-81 2 Gertz MA et al.: Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol 2015; 12(2): 91-102 3 González-López E et al.: Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36(38): 2585-94 4 Damy T et al.: Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J 2016; 37(23): 1826-34 5 González-López E et al.: Clinical characteristics of wild-type transthyretin cardiac amyloidosis: Disproving myths. Eur Heart J 2017; 38(24): 1895-904 6 Banypersad SM et al.: Updates in cardiac amyloidosis: A review. J Am Heart Assoc 2012; 1(2): e000364
7 González-López E et al.: Diagnosis and treatment of transthyretin cardiac amyloidosis. progress and hope. Rev Española Cardiol (English Ed.) 2017; 70(11): 991-1004 8 Haaf P et al.: Cardiac T1 mapping and extracellular volume (ECV) in clinical practice: A comprehensive review. J Cardiovasc Magn Reson 2016; 18(1): 89 9 Gillmore JD et al.: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation2016; 133(24):2404-12 10 Gertz MA et al.: Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol 2015; 66(21): 2451-66 11 Maurer MS et al.: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379(11): 1007-16
Das könnte Sie auch interessieren:
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...

Immunsuppression und Infektion: Durchimpfen so gut wie möglich
Autoimmunerkrankungen sind mit erhöhtem Infektionsrisiko verbunden, das durch immunsupprimierende Therapien weiter verstärkt wird. Impfungen sind also in der betroffenen Patient: ...