
Erste internationale Leitlinie zu allen Formen von Kardiomyopathien
Die ESC hat die erste internationale Leitlinie zur Behandlung von Kardiomyopathien (CMP) herausgegeben, die alle CMP-Arten enthält.1 Sie soll das Bewusstsein für die Ätiologie der CMP-Formen schärfen und umfasst den Phänotyp-orientierten diagnostischen Zugang, das allgemeine Management sowie jeweilige Besonderheiten. Im Fokus stehen Empfehlungen zur multimodalen Bildgebung, zum genetischen Testen, zum Familienscreening, zum Umgang mit dem CMP-Risiko, zur manifesten CMP und zum Risiko eines „sudden cardiac death“ (SCD).
Keypoints
-
Eine CMP-Diagnose sollte die Ursachensuche anhand des Phänotyp-basierten diagnostischen Pfades zur Folge haben.
-
Für das CMP-Management sind die entsprechenden Leitlinien für Herzinsuffizienz, Rhythmus und Devices heranzuziehen, bei manchen Phänotypen/Ätiologien sind jedoch Besonderheiten zu beachten.
-
Für die frühzeitige Implantation eine ICDs aufgrund von Varianten in Risikogenen gewinnt das genetische Testen zur Reevaluierung des Arrhythmiesierungsrisikos immer mehr an Bedeutung.
-
Eine genetische Kaskadentestung sollte bei Verdacht auf eine familiäre CMP bei sicherem genetischem Hintergrund (P/LP-Variante) Verwandten und Kindern angeboten werden.
Die neue ESC-Leitlinie 2023 zum CMP-Management ist eine wertvolle Ergänzung zu bisherigen Leitlinien. Nachdem die Leitlinie neu ist, sind dies die meisten Empfehlungen ebenso. Eine Ausnahme bilden Empfehlungen zu hypertrophen Kardiomyopathien (HCM), für die bereits 2014 eine ESC-Leitlinie existierte.2 Diese wurde in die aktuelle Leitlinie integriert und entsprechend den neuesten Erkenntnissen überarbeitet. Da es zu den meisten spezifischen CMP nur wenige randomisierte kontrollierte Studien (RCTs) gibt, basieren die meisten Empfehlungen auf Expertenmeinungen – also Evidenzgrad C. Einzelne spezifische CMP-Ätiologien werden nicht ausführlich beschrieben, sondern es wird auf die Literatur verwiesen. In der Leitlinie finden sich wichtige Neuerungen und eine adaptierte Strukturierung (Abb. 1).
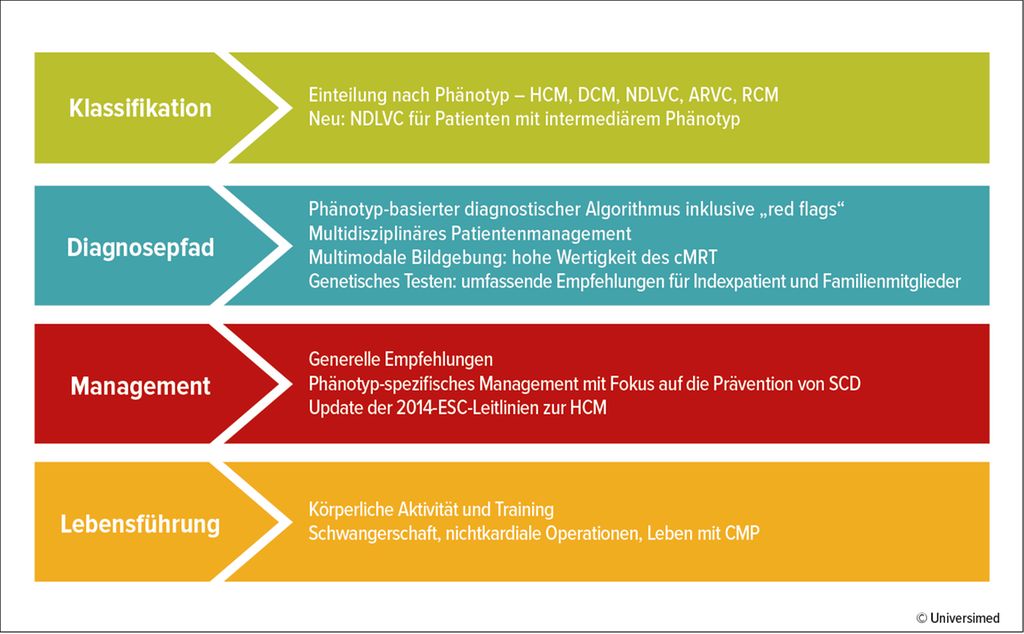
Abb. 1: Überblick über die ESC-Leitlinie 2023 zum Management der Kardiomyopathien (nach Arbelo E et al. 2023)1
Welche Kardiomyopathien werden in der Leitlinie behandelt?
Der Begriff Kardiomyopathie bezeichnet allgemein eine Störung des Myokards, bei der es strukturelle oder funktionelle Abnormalitäten gibt.3 Historisch wurde eine CMP, die durch eine Ischämie, also zumeist eine koronare Herzerkrankung (KHK), verursacht wurde, von der engeren Bezeichnung CMP ausgeschlossen. Im Laufe der Jahre wurden regelmäßig neue Definitionen und Klassifikationsschemata von der Weltgesundheitsorganisation sowie von nationalen und internationalen Fachgesellschaften bereitgestellt. Die aktuelle Leitlinie fokussiert auf CMP, die nicht ausreichend durch eine KHK, Hypertonie, valvuläre Herzerkrankungen oder angeborene Herzfehler erklärt werden können. Es können auch mehrere Ursachen gleichzeitig vorliegen. Die Ätiologie zu kennen ist wichtig, da dies einen Unterschied in der Patientenaufklärung, der Prognoseabschätzung und im Management macht, insbesondere bei der Bewertung des Risikos für einen SCD und in speziellen Formen auch bei der Therapie. Daher sollte die Diagnose CMP die Suche nach einem Auslöser anstoßen. Bei genetischer CMP spielen die Früherkennung, Risikobewertung und Therapie betroffener Familienmitglieder sowie die Wahrscheinlichkeit für die Weitergabe an Nachkommen bei der Familienplanung eine entscheidende Rolle.
Phänotypen der Kardiomyopathien
Seit den frühen 1960er-Jahren wird zur Klassifikation von CMP ein pragmatischer, deskriptiver Ansatz verfolgt, der sich auf die Morphologie konzentriert. Gemäß dem ESC-Konsensusdokument von 2008 sollen die vier Phänotypen HCM, dilatative CMP (DCM), arrhythmogene rechtsventrikuläre CMP (ARVC) und restriktive CMP (RCM) sowie nichtklassifizierte CMP auch nach familiärer Häufung in familiär/genetisch und nichtfamiliär/nichtgenetisch unterteilt werden.3 Die MOGES-Klassifikation von 2014 aus dem amerikanischen Raum versucht, die CMP ganzheitlicher zu erfassen, indem sie den Phänotyp (M=Morphologie), die genetische Beteiligung (G=genetisches Vererbungsmuster), die Beteiligung anderer Organsysteme (O=Organbeteiligung), den Krankheitsstatus (S=Stadium) und die Ätiologie (E=Ursache) als Klassifikationskriterien berücksichtigt.4 Die aktuelle Leitlinie übernimmt die vier Phänotypen und ergänzt sie um den neuen fünften Phänotyp „nichtdilatierte linksventrikuläre Kardiomyopathie“ (NDLVC). NDLVC umfasst Patienten mit isolierter linksventrikulärer Dysfunktion ohne Narbenbildung oder Patienten mit nichtischämischer Narbenbildung unabhängig von einer systolischen Dysfunktion. Dadurch können intermediäre Phänotypen erfasst werden, die trotz Vorliegen einer Myokarderkrankung nicht den Definitionen der anderen Klassen entsprechen (Tab. 1).

Tab. 1: Pragmatischer Ansatz zur ersten Klassifikation der Kardiomyopathien: der Phänotyp (nach Arbelo E et al. 2023)1
Der diagnostische Pfad wird durch den Phänotypen bestimmt
Die Festlegung des CMP-Phänotyps ist der erste Schritt im diagnostischen Prozess, um die Ätiologie festzulegen. Patienten mit einer CMP können wenig bis keine kardialen Symptome aufweisen; viele nichtkardiale Symptome können wichtige Hinweise auf die Ätiologie geben. Auffälligkeiten im EKG, Herzrhythmusstörungen, SCD, manifeste Herzinsuffizienz (HI), auffällige Ergebnisse beim Familienscreening oder Multiorganerkrankungen können Grund zur Erstvorstellung sein.
Bei Verdacht auf eine CMP oder bei manifester CMP wird ein multiparametrischer Ansatz mit klinischer Bewertung, Stammbaumanalyse, EKG, Holter-Monitoring, Labortests und multimodaler Bildgebung empfohlen (IC). Die Stammbaumanalyse sollte 3–4 Generationen umfassen, um bei der Diagnosestellung den Vererbungsmodus zu bestimmen und Familienmitglieder mit erhöhtem Risiko zu identifizieren (IC).
Ein wichtiger diagnostischer Bestandteil ist die multimodale Bildgebung. Neben der Echokardiografie als Standarduntersuchung spielt die kardiale Magnetresonanztomografie mit Kontrastmittel (cMRT) eine entscheidende Rolle bei der Abklärung. Die cMRT wird bei allen Patienten bei Erstvorstellung empfohlen (IB) und kann dazu beitragen, den Verlauf der Krankheit einzuschätzen oder das Ansprechen auf die Therapie zu bewerten (IIaC). Die cMRT kann auch zur Früherkennung bei Personen mit genetisch positivem und phänotypisch negativem Status sowie bei phänotypisch negativem, aber unbekanntem genetischem Status eingesetzt werden (IIaB und IIbC). In speziellen Verdachtsfällen oder zum Ausschluss anderer Diagnosen können eine kardiale Computertomografie (cCT), eine PET-CT oder eine Endomyokardbiopsie durchgeführt werden.
Ein Schwerpunkt der Leitlinie liegt auf der genetischen Diagnostik. CMP sind durch eine ausgeprägte genetische und allelische Heterogenität gekennzeichnet. D.h., dass verschiedene Varianten in verschiedenen Genen den gleichen Krankheitsphänotyp verursachen können. Es ist wichtig zu wissen, dass nicht alle Personen mit einer kausalen genetischen Mutation eine CMP entwickeln. Bei jenen, die eine entwickeln, gibt es eine breite Streuung in Bezug auf das Alter und die Schwere der Erkrankung.5,6 Sowohl bei DCM als auch bei HCM scheint häufig ein polygener Hintergrund mit komplexer Vererbung vorzuliegen. Anstelle einer seltenen Mutation mit großem Effekt liegt hier eher eine Mitvererbung mehrerer Suszeptibilitätsgene mit variablem Effekt vor. In Zukunft könnten polygene Risikoscores die kumulative Wirkung vieler Varianten mit geringem Effekt besser abbilden und über das Management informieren.
Bei mehreren CMP-Formen, die früher als sekundär angesehen wurden, wie z.B. bei der peripartalen Kardiomyopathie, der kardiotoxischen Kardiomyopathie und bei der Myokarditis, wurde kürzlich eine genetische Rolle nachgewiesen. Dies führte zur Postulierung der „Second-Hit-Theorie“.
Die Leitlinie enthält eine Tabelle mit einer Auflistung aller Gene, deren Varianten mit monogenen, nichtsyndromischen CMP in Verbindung gebracht wurden, wobei der geschätzte Beitrag zur Manifestation der jeweiligen CMP angeführt wird. Die genetische Testung wird bei Patienten empfohlen, die die Diagnosekriterien für eine CMP erfüllen und für die ein Nutzen für sich selbst oder ihre Familienmitglieder zu erwarten ist (IB). Eine genetische Testung kann auch zur weiteren Abklärung einer grenzwertigen CMP in Betracht gezogen werden (IIbC). Eine Kaskadentestung mit vorheriger und nachfolgender Beratung wird für Familien empfohlen, bei denen eine genetische CMP mit einer „pathogenic/likely pathogenic“Variante (P/LP-Variante) gesichert wurde, beginnend bei Verwandten ersten Grades mit sequenzieller Weiterführung (IB). Das Testen von Verwandten bei einer Variante mit unbekannter Signifikanz (VUS) in der Familie kann ebenfalls erwogen werden, um die Relevanz der Variante festzustellen (IIaC). Wenn bei einem Patienten mit CMP keine P/LP-Variante festgestellt wird, sollten phänotypisch negative Verwandte keinem genetischen Test unterzogen werden (IIIC).
Die Leitlinie gibt auch eine Empfehlung bezüglich der genetischen Testung bei Kindern ab, wobei der Zeitpunkt je nach Setting variieren kann. Die Bedeutung des Übergangs von der pädiatrischen zur Erwachsenenbetreuung wird betont.
Management von Kardiomyopathie
Im Abschnitt zum generellen Management werden verschiedene Strategien diskutiert, wie die Erfassung von Symptomen, die Behandlung der HI sowie atriale und ventrikuläre Arrhythmien, Device-Therapien und klinische Kontrollen.
Die Behandlung der HI richtet sich nach den allgemeinen HI-Leitlinien, unabhängig von der Ätiologie.7 Die kardiale Amyloidose und die restriktive CMP erfordern ein spezielles Management, bei dem eine Euvolämie im Mittelpunkt steht.8 Eine besondere Gruppe sind Patienten mit asymptomatischer linksventrikulärer Dysfunktion, was frühen Formen einer CMP entsprechen kann. Eine HI-Therapie kann bei NDLVC in Betracht gezogen werden, da sie das Remodeling positiv beeinflusst (IIbC). Außerdem könnten Biomarker, wie natriuretische Peptide, Patienten aufdecken, die von einer frühzeitigen neurohumoralen Blockade profitieren können.9 Bei genetisch positiven und phänotypisch negativen Personen gibt es keinen Hinweis auf den Nutzen einer medikamentösen Therapie.
Bezüglich der kardialen Komorbidität Vorhofflimmern ist bekannt, dass einige CMP, wie die HCM und die kardiale Amyloidose, mit einer höheren Schlaganfallrate assoziiert sind.10 In solchen Fällen sollte unabhängig vom CHA2DS2-VASc-Score eine therapeutische Antikoagulation eingeleitet werden (IIaC).11 Eine Frequenzkontrolle sollte bei allen Patienten mit Vorhofflimmern und CMP erfolgen.12 Betablocker sind die erste Wahl, alternativ kann Digoxin in Betracht gezogen werden. Verapamil oder Diltiazem sollten nur bei einer linksventrikulären Auswurffraktion (LVEF) über 40% verwendet werden.
Für ventrikuläre Arrhythmien gelten die aktuellen ESC-Leitlinien für das Management von ventrikulären Arrhythmien und die Prävention des SCD. Bei der Implantation eines implantierbaren Kardioverterdefibrillators (ICD) als Primärprävention sollten einige Besonderheiten beachtet werden. Das Arrhythmierisiko von Patienten mit CMP sollte regelmäßig alle 1–2 Jahre oder bei Veränderungen im klinischen Status reevaluiert werden (IC). Dabei wird die Verwendung von Risikoscores und SCD-Algorithmen empfohlen. Diese Abschätzung könnte die klinische Schwelle für eine ICD-Implantation senken, wobei insbesondere betont wird, dass die Entscheidung gemeinsam mit dem Patienten nach ausführlicher Aufklärung („shared decision making“) getroffen werden muss.
Patienten mit einer CMP sollten alle 1–2 Jahre einer klinischen Untersuchung mit EKG und Echokardiografie unterzogen werden (IC) oder wenn sich die Symptome unerwartet oder signifikant verändern (IC). Eine kardiale cMRT kann alle 2–5 Jahre, oder bei schnell fortschreitender Erkrankung auch häufiger, durchgeführt werden. Neue Empfehlungen gibt es auch zum Management von Verwandten eines Patienten mit CMP je nach Vorhandensein und Ergebnis eines genetischen Tests (Abb. 2).

Abb. 2: ESC-Algorithmus zum Familienscreening und Follow-up bei Familienmitgliedern (nach Arbelo E et al., 2023)1
Management von CMP-Phänotypen
Die Empfehlungen für HCM sind ein Update der ESC-Leitlinien aus dem Jahr 2014. In Bezug auf die pharmakologische Therapie der HCM mit linksventrikulärer Ausflusstraktobstruktion (LVOTO) wurde Mavacamten, ein kardialer Myosin-ATPase-Inhibitor, aufgrund der Ergebnisse der Studien EXPLORER-HCM und VALOR-HCM in die Leitlinien aufgenommen.13–15 Mavacamten kann als Zweitlinientherapie in Betracht gezogen werden (IIaA), wobei die Substanz mit Betablockern und Kalziumantagonisten kombiniert werden sollte, nicht aber mit Disopyramid. Bei Unverträglichkeit oder Kontraindikationen gegenüber anderen Medikamenten kann eine Monotherapie mit Mavacamten in Erwägung gezogen werden (IIaB). Die Dosissteigerung sollte mittels Echokardiografie überwacht werden. Für die Prävention des SCD empfehlen die Leitlinien eine Risikoevaluierung mittels Risikorechnern als ersten Schritt.16–18 Bei einem 5-Jahres-Risiko von >6% sollte eine primärprophylaktische ICD-Implantation erwogen werden (IIaB). Bei 4–6% kann diese in Betracht gezogen werden und bei <4% können ein hohes „Late Gadolinium Enhancement“ (LGE) >15% oder eine LVEF <50% als zusätzlicher Risikofaktor in der Entscheidung mitberücksichtigt werden (IIbB).
Für das Management der DCM gelten die Leitlinien der HI und der Devices.7,19 Bestimmte Mutationen erhöhen das Arrhythmierisiko unabhängig von der LVEF.20–25 Bei diesen Patienten mit Hochrisikogenen (z.B. LMNA, FLCN truncating variants, TMEM43, PLN, DSP, RBM20) könnte die Implantation eines primärprophylaktischen ICD bereits bei einer LVEF >35% in Erwägung gezogen werden, wenn andere Risikofaktoren wie nsVTs (nichtanhaltende ventrikuläre Tachykardien), ventrikuläre Ektopien oder ausgeprägtes LGE im cMRT vorhanden sind. Es laufen Studien, die die Bedeutung von LGE in der DCM im Zusammenhang mit SCD genauer untersuchen (NCT04558723, NCT03993730).
Patienten mit NDLVC-Phänotyp sollten der gleichen Diagnostik unterzogen werden wie jene mit anderen CMP-Phänotypen. Die häufigsten assoziierten Gene sind DSP, FLNC, DES, LMNA und PLN. Die Empfehlungen zur ICD-Implantation entsprechen denen der DCM.
Praxistipp
Der SCD-Risikorechner findet sich unter: https://qxmd.com/calculate/calculator_303/hcm-risk-scdDie ARVC hat eine altersabhängige Penetranz und ist durch eine hohe klinische und genetische Variabilität charakterisiert. Eine ARVC sollte bei jungen Erwachsenen mit Palpitationen, Synkopen oder nach Überleben eines SCD suszipiert werden. Häufig präsentiert sich die ARVC mit Linksschenkelblock (LSB), ventrikulären Extrasystolen (VES) oder ventrikulären Tachykardien (VT), manchmal mit einer Dilatation des rechten Ventrikels. Die cMRT kann durch Darstellung des „fibro-fatty replacement“ die Involvierung des linken Ventrikels abschätzen. Betablocker sind erste Wahl bei der Kontrolle von VES, nsVT und VT (IC). Obwohl die Erkrankung selten ist, wird eine ARVC häufig als Ursache von SCD in Registern genannt.26 Der 2019 überarbeitete „ARVCRisk Calculator“ sollte zur Risikoabschätzung herangezogen werden (IIaB). Auch „high-risk features“ sollte Bedeutung beigemessen werden (IIaB).27–30
Die restriktive CMP ist die CMP mit der schlechtesten Prognose. Mehr als 75% der überlebenden Patienten präsentieren sich mit HI. Ohne Herztransplantation führt die Erkrankung wenige Jahre nach Diagnosestellung zum Tod.31–33 Die pulmonalen Drücke können, auch ohne klinische Veränderungen, schnell steigen, was bei der zeitlichen Planung einer Transplantation berücksichtigt werden muss.
Management in besonderen Situationen und Lebensführung
In den weiteren Abschnitten beinhaltet die Leitlinie Empfehlungen zu körperlicher Aktivität und Training unddas Management in der Schwangerschaft oder bei nichtkardialen Operationen und diskutiert weitere Aspekte der Lebensführung.
Literatur:
1 Arbelo E et al.: 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J 2023; 44(37): 3503-626 2 Elliott PM et al.: 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39): 2733-79 3 Elliott P et al.: Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008; 29(2): 270-6 4 Arbustini E et al.: The MOGE(S) classification of cardiomyopathy for clinicians. J Am Coll Cardiol 2014; 64(3): 304-18 5 Lorenzini M et al.: Penetrance of hypertrophic cardiomyopathy in sarcomere protein mutation carriers. J Am Coll Cardiol 2020; 76(5): 550-9 6 Tadros R et al.: Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet 2021; 53(2): 128-34 7 McDonagh TA et al.: 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2021; 42(36): 3599-726 8 Oghina S et al.: The impact of patients with cardiac amyloidosis in HFpEF trials. JACC Heart Fail 2021; 9(3): 169-78 9 Mureddu GF et al.: Evaluation of different strategies for identifying asymptomatic left ventricular dysfunction and pre-clinical (stage B) heart failure in the elderly. Results from PREDICTOR, a population based-study in central Italy. Eur J Heart Fail 2013; 15(10): 1102-12 10 Mizia-Stec K et al.: Atrial fibrillation, anticoagulation management and risk of stroke in the cardiomyopathy/myocarditis registry of the EURObservational research programme of the European Society of Cardiology. ESC Heart Fail 2020; 7(6): 3601-9 11 Fauchier L et al.: Ischemic stroke in patients with hypertrophic cardiomyopathy according to presence or absence of atrial fibrillation. Stroke 2022; 53(2): 497-504 12 Hindricks G et al.: 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): the task force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur Heart J 2021; 42(5): 373-498 13 Olivotto I et al.: Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020; 396(10253): 759-69 14 Desai MY et al.: Myosin inhibition in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy. J Am Coll Cardiol 2022; 80(2): 95-108 15 Desai MY et al.: Dose-blinded myosin inhibition in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy: outcomes through 32 weeks. Circulation 2023; 147(11): 850-63 16 O’Mahony C et al.: A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30): 2010-20 17 Vriesendorp PA et al.: Validation of the 2014 European Society of Cardiology guidelines risk prediction model for the primary prevention of sudden cardiac death in hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2015; 8(4): 829-35 18 O’Mahony C et al.: International external validation study of the 2014 European Society of Cardiology Guidelines on sudden cardiac death prevention in hypertrophic cardiomyopathy (EVIDENCE-HCM). Circulation 2018; 137(10): 1015-23 19 Glikson M et al.: 2021 ESC Guidelines on cardiac pacing and cardiac resynchronization therapy. Europace 2022; 24(1): 71-164 20 Kumar S et al.: Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J Am Coll Cardiol 2016; 68(21): 2299-307 21 Verstraelen TE et al.: Prediction of ventricular arrhythmia in phospholamban p.Arg14del mutation carriers-reaching the frontiers of individual risk prediction. Eur Heart J 2021; 42(29): 2842-50 22 Gigli M et al.: Phenotypic expression, natural history, and risk stratification of cardiomyopathy caused by filamin C truncating variants. Circulation 2021; 144(20): 1600-11 23 Akhtar MM et al.: Association of left ventricular systolic dysfunction among carriers of truncating variants in filamin C with frequent ventricular arrhythmia and end-stage heart failure. JAMA Cardiol 2021; 6(8): 891-901 24 Hodgkinson KA et al.: Long-term clinical outcome of arrhythmogenic right ventricular cardiomyopathy in individuals with a p.S358L mutation in TMEM43 following implantable cardioverter defibrillator therapy. Circ Arrhythm Electrophysiol 2016; 9(3): e003589 25 Hey TM et al.: Pathogenic RBM20-variants are associated with a severe disease expression in male patients with dilated cardiomyopathy. Circ Heart Fail 2019; 12(3): e005700 26 Finocchiaro G et al.: Etiology of sudden death in sports: insights from a United Kingdom regional registry. J Am Coll Cardiol 2016; 67(18): 2108-15 27 Protonotarios A et al.: Importance of genotype for risk stratification in arrhythmogenic right ventricular cardiomyopathy using the 2019 ARVC risk calculator. Eur Heart J 2022; 43(32): 3053-67 28 Bosman LP et al.: Comparing clinical performance of current implantable cardioverter-defibrillator implantation recommendations in arrhythmogenic right ventricular cardiomyopathy. Europace 2022; 24(2): 296-305 29 Cadrin-Tourigny et al.: A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2019; 40(23): 1850-8 30 Bosman LP et al.: Predicting arrhythmic risk in arrhythmogenic right ventricular cardiomyopathy: a systematic review and meta-analysis. Heart Rhythm 2018; 15(7): 1097-107 31 Mori H et al.: Outcomes of restrictive cardiomyopathy in Japanese children - a retrospective cohort study. Circ J 2022; 86(12): 1943-9 32 Ammash NM et al.: Clinical profile and outcome of idiopathic restrictive cardiomyopathy. Circulation 2000; 101(21): 2490-6 33 Mogensen J, Arbustini E: Restrictive cardiomyopathy. Curr Opin Cardiol 2009; 24(3): 214-20
Das könnte Sie auch interessieren:

Mechanische Kreislaufunterstützung im Infarkt-bedingten kardiogenen Schock
Der Infarkt-bedingte kardiogene Schock (AMI-CS) ist trotz der enormen Fortschritte in der interventionellen Versorgung des akuten Myokardinfarktes in den vergangenen Jahrzehnten ...
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...