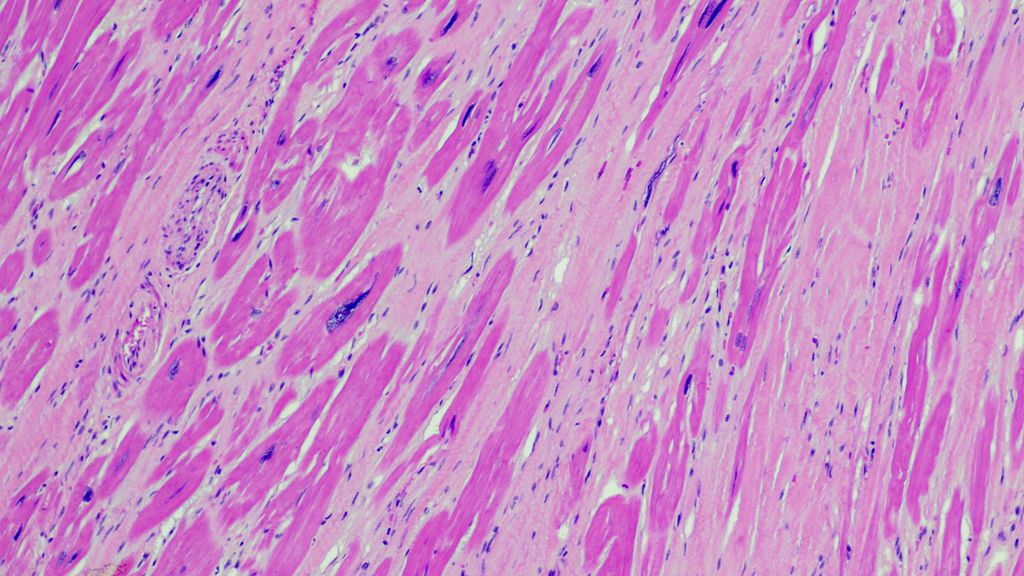
Hypertrophe Kardiomyopathie
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die hypertrophe Kardiomyopathie (HCM) ist eine der häufigsten genetisch vererbbaren kardialen Erkrankungen. Eine oft vorhandene Einengung des linksventrikulären Ausflusstraktes bedarf meist unterschiedlicher Therapiekonzepte, ebenso kann es zum Auftreten maligner Rhythmusstörungen kommen. Die kardiale Amyloidose stellt eine Sonderform der HCM dar, die oft mit einer schlechten Prognose verbunden ist und daher bereits frühzeitig erkannt werden sollte.
Keypoints
-
Der hypertrophen Kardiomyopathie liegt oft eine genetische Mutation zugrunde.
-
Neben bereits etablierten Behandlungsoptionen folgt 2023 die Zulassung von Mavacamten.
-
Die Leichtketten- (AL) und die Transthyretin(ATTR)-Amyloidose stellen Sonderformen dar.
-
Frühzeitige Erkennung sowie Therapieinitiierung sind essenziell.
Die hypertrophe Kardiomyopathie (HCM) ist definiert als interventrikuläre Septumdicke ≥15 mm ohne fassbare Ursache (z.B. langjährige Hypertonie, hämodynamisch relevante Vitien u.a.).1,2 Die häufigste Form stellt die asymmetrische/vorrangig das Septum betreffende HCM dar, jedoch sind auch rein apikale bzw. Mischformen möglich. In 40–60% der Fälle liegt eine Genmutation (autosomal dominanter Erbgang) vor, allerdings können auch diverse Speichererkrankungen oder neurologische Erkrankungsbilder in einer HCM resultieren (Abb. 1).

Abb. 1: Hypertrophe Kardiomyopathie bei diversen Speichererkrankungen oder neurologischen Erkrankungsbildern
Ein Druckgradient ≥30mmHg beschreibt eine relevante Obstruktion, die meist den linksventrikulären Ausflusstrakt (LVOT) betrifft, jedoch gelegentlich auch in mesoventrikulären Abschnitten vorkommen kann. Im Rahmen einer signifikanten LVOT-Obstruktion kommt es zu einer anterior gerichteten Bewegung der Mitralklappensegel (SAM-Phänomen) mit einer konsekutiv meist deutlich ausgeprägten Mitralklappeninsuffizienz.
Die Prävalenz der HCM beträgt in etwa 1:500, wobei ca. 2/3 der Patienten eine relevante Obstruktion aufweisen (HOCM).
Etablierte Therapieoptionen der hypertrophen Kardiomyopathie
Die Behandlungsoptionen von Patienten mit relevanter LVOT-Obstruktion bestehen aus drei Säulen, die auf einem dreistufigen Fundament stehen: Euvolämie anstreben, Spitzenbelastungen vermeiden sowie starke Blutdruckschwankungen verhindern. Im Folgenden werden die drei Säulen näher beleuchtet:
Medikamentös
Hier haben sich sowohl kardioselektive Betablocker sowie Kalziumkanalblocker vom Nicht-Dihydropyridin-Typ (Verapamil, Diltiazem) etabliert. Ihr gemeinsames Ziel ist es den O2-Bedarf zu senken, den „Postexercise“-Gradienten zu reduzieren sowie die diastolische Füllungsphase zu prolongieren. Sollte es hierdurch zu keiner klinischen Besserung kommen bzw. der Gradient weiterhin hoch sein (≥50mmHg), können entsprechende Interventionen in Erwägung gezogen werden.
Alkoholablation
Bei diesem Eingriff, der in der Literatur als TASH („transcoronary ablation of septal hypertrophy“) oder auch PTSMA („percutaneous transluminal septal myocardial ablation“) bezeichnet wird, wird im Rahmen einer Koronarangiografie der 1. septale Ast des Vorderwandgefäßes (LAD) sondiert und ein Ballon inflatiert. Durch einen kleinen Katheter, der durch den Ballon hindurchgeht, wird Ethanol injiziert, der durch direkte Zytotoxizität das versorgende Septumareal zur Vernarbung bringen und somit den LVOT-Gradienten reduzieren soll (Abb. 2).

Abb. 2: Schematische Darstellung einer TASH („transcoronary ablation of septal hypertrophy“)
Chirurgische Myektomie
Bei der chirurgischen Myektomie (nach Morrow) wird durch einen meist trans-aortalen Zugang der entsprechende Septumwulst reseziert. Manchmal ist jedoch aufgrund zusätzlich fibrosierter Sehnenfäden und/oder struktureller Abnormalitäten der Mitralklappe eine Erweiterung des Eingriffs notwendig.
Bezüglich der Vergleichbarkeit beider Methoden existieren bislang keine prospektiv randomisierten Studien, weshalb eine individuelle,patientenorientierte Risiko-Nutzen-Abwägung in entsprechenden Zentren erfolgen sollte. Während bei der Alkoholablation die „Redo“-Rate deutlich erhöht ist und bis zu 20% der Patienten eine permanente Schrittmacherimplantation benötigen, stellt die chirurgische Myektomie die deutlich invasivere Behandlungsoption mit längerem Spitalsaufenthalt dar und kann zu ventrikulären Septumdefekten führen. Allerdings sollte sie bei gleichzeitig bestehenden strukturellen Problemen an Sehnenfäden bzw. der Mitralklappe (und auch bei Notwendigkeit der Sanierung anderer Klappen) bevorzugt werden.4
Risikostratifizierung hinsichtlich des plötzlichen Herztodes
Das größte Risiko bei HCM-Patienten besteht im Auftreten maligner Rhythmusstörungen. Deshalb ist dieses Risiko in regelmäßigen Abständen zu evaluieren, um ggf. eine primär prophylaktische Implantation eines Defibrillators initiieren zu können. Hierzu wird der HCM-Risikorechner der europäischen Leitlinien herangezogen, der anhand folgender Parameter das Risiko eines plötzlichen Herztodes innerhalb der nächsten 5 Jahre berechnet: Alter, maximale Wanddicke, Größe des linken Vorhofs, maximaler LVOT-Gradient, familiäre Anamnese des plötzlichen Herztodes, unerklärte Synkope bzw. nichtanhaltende ventrikuläre Tachykardien in der Anamnese.1 Zusätzliche Risikofaktoren sind das Vorhandensein von apikalen Aneurysmen bzw. ein Narbenareal ≥15% des gesamten linken Ventrikels, die jedoch erst in die amerikanischen Richtlinien Einzug gefunden haben.2
Als UltimaRatio ist bei therapierefraktären Patienten immer an eine Herztransplantation zu denken.
Ausblick
Für das erste direkte Therapeutikum der HCM, Mavacamten, steht die Zulassung in Österreich Anfang 2023 direkt bevor. Der niedermolekulare Myosinmodulator soll die bei HCM vermehrt ausgeprägten Aktin-Myosin-Brücken inaktivieren und somit der Hyperkontraktilität entgegenwirken. Rezent publizierte Studien wie EXPLORER-HCM sowie VALOR-HCM haben bereits eindrucksvolle Ergebnisse geliefert.5,6
Kardiale Amyloidose
Pathophysiologie und Epidemiologie
Bei dieser Erkrankung lagern sich fehlgefaltete Proteine im Extrazellularraum des Myokards an und führen so zur Herzmuskelhypertrophie. Obwohl über 30 Proteine eine Amyloidose verursachen können, sind aus kardiologischer Sicht im Wesentlichen nur 2 in der klinischen Routine relevant: Zum einen sind dies Leichtketten (AL), welche von einem Plasmazellklon produziert werden,zum anderen Transthyretin (ATTR), welches hauptsächlich von der Leber synthetisiert wird. Bei der ATTR-Amyloidose wiederum gilt es noch zwischen einer altersassoziierten Wildtypamyloidose (ATTRwt) und einer durch Punktmutationen verursachten genetischen (ATTRv) Amyloidose zu unterscheiden (Abb. 3).8 Epidemiologisch zeigte sich in den letzten Jahren eine deutliche Zunahme der Prävalenz und Inzidenz der kardialen Amyloidose, wobei diese Entwicklung vor allem auf einer Zunahme der ATTRwt-Amyloidose beruht.
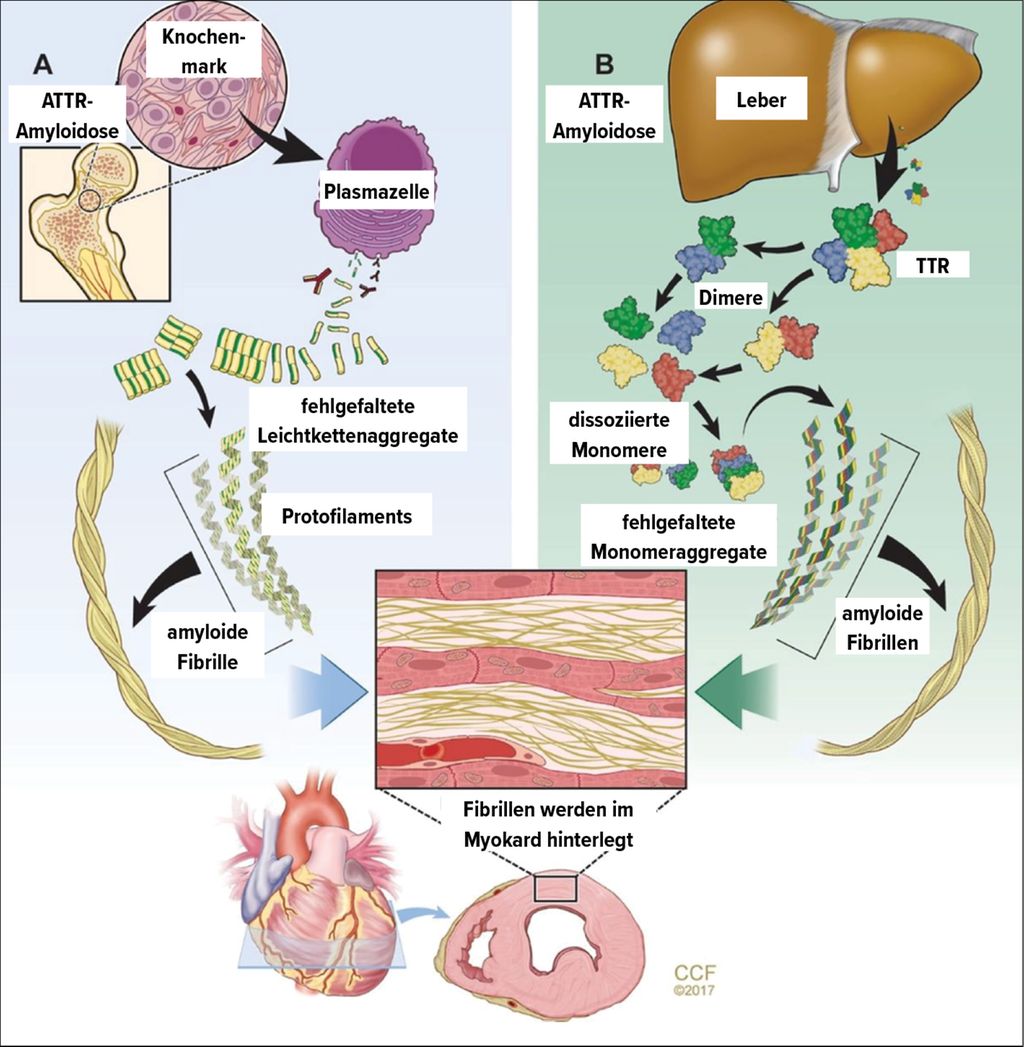
Abb. 3: Differierende Pathophysiologie AL- und der ATTR-Amyloidose (aus Rubin J, Maurer MS: Annu Rev Med 2020; 71: 203-19, mit Permission des Verlags)
Diagnose
Durch die zunehmende Bedeutung dieser Erkrankung wurde im Jahr 2020 ein österreichischer Konsensus zu Diagnose und Therapie veröffentlicht.9
1. Schritt
Das Wichtigste ist, überhaupt an eine kardiale Amyloidose zu denken und diese Erkrankung in das differenzialdiagnostische Denken aufzunehmen. Studien konnten in den letzten Jahren Populationen mit hohem Risiko herausarbeiten:10,11
-
5% bei ≥70-Jährigen (Autopsie-Populationsstudie)
-
25% bei ≥85-Jährigen (Autopsie-Populationsstudie)
-
20% bei Patienten mit HFpEF
-
15% bei Patienten mit Aortenklappenstenose (vor allem TAVI und [paradoxer] „low-flow-low-gradient“ Aortenstenose)
-
5% bei Patienten mit hypertropher Kardiomyopathie
-
weiße Männer höheren Alters mit Herzinsuffizienz/-hypertrophie
Obwohl die ATTR-Amyloidose die numerisch wichtigste Amyloidoseform darstellt, ist es von größter Wichtigkeit, eine AL-Amyloidose früh zu erkennen, da hier das therapiefreie Überleben (auch bei sehr jungen Patienten) lediglich 3–6 Monate umfasst.
2. Schritt
Der zweite Schritt im Diagnosealgorithmus ist die Durchführung einer Serum-/Harnanalyse auf das Vorliegen eines Paraproteins (AL-Amyloidose) und einer Ganzkörperknochenszintigrafie, diese wird auch auch DPD-Scan genannt (ATTR-Amyloidose).
Wenn beide Untersuchungen unauffällig ausfallen, ist eine Amyloidose nahezu sicher auszuschließen.
Wenn lediglich der DPD-Scan positiv ausfällt (cave: Perugini-Grad ≥2) und im Serum/Harn kein Paraprotein zu finden ist, kann die ATTR-Amyloidose bereits nichtinvasiv diagnostiziert werden. Bei anderen Befundkonstellationen muss eine weiterführende Diagnostik mittels Organbiopsien erfolgen (Tab.1).

Tab. 1: Diagnostische Schritte zum Ausschluss bzw. zur Bestätigung von Amyoloidoseformen
Therapie
Die Therapie der kardialen Amyloidose fußt auf den 3 Säulen der supportiven Therapie, der Amyloidose-spezifischen medikamentösen Therapie und der Organtransplantation.
Die supportive Therapie der kardialen Amyloidose umfasst vor allem das Flüssigkeitsmanagement mittels Diuretika sowie die Behandlung diverser Rhythmusstörungen.
Bei der spezifischen Therapie stehen nun sowohl für die kardiale AL als auch für die ATTR-(wt & v)Amyloidose spezifische Therapien zur Verfügung, welche zu einer signifikanten Verlängerung des Überlebens betroffener Patienten führen (Abb. 4 und 5).12,13

Abb. 4: Kaplan-Meier-Kurve zur signifikanten Verlängerung des Überlebens durch Daratumumab bei Patienten mit kardialer Amyloidose (nach Kastritis et al. 2021)12

Abb. 5: Signifikante Verlängerung des Gesamtüberlebens durch Tafamidis bei ATTR-Amyloidose(modifiziert nach Maurer et al. 2018)13
Jedoch sei an dieser Stelle nochmals auf die entscheidende Rolle einer möglichst frühen Diagnosestellung als Basis für eine erfolgreiche medikamentöse Therapie hingewiesen.
Zusammenfassung
Die kardiale Amyloidose hat sich von einer „Orphan Disease“ zu einer klinisch relevanten Form der HCM entwickelt, insbesondere, da nun seit Kurzem Amyloidose-spezifische medikamentöse Therapien zur Verfügung stehen, welche das Überleben betroffener Patienten signifikant verlängern.
Literatur:
1 Elliot PM et al.: 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39): 2733-79 2 Ommen SR et al.: 2020 AHA/ACC Guidelines for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: Executive summary: A report of American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020; 142(25): e533-57 3 www.genesiscare.com 4 Arevalos V et al.: Alcohol septal ablation: An option on the rise in hypertrophic cardiomyopathy. J Clin Med 2021; 10(11): 2276 5 Olivotto I et al.: Mavacamten for the treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020; 396(10253): 759-69 6 Desai MY et al.: Myosin inhibition in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy. J Am Coll Cardiol 2022; 80(2): 95-108 7 Donnelly JP et al.: Cardiac amyloidosis: An update on diagnosis and treatment. Cleve ClinJMed 2017; 84: 12-26 8 Rubin J, Maurer MS: Cardiac Amyloidosis: Overlooked, Underappreciated, and Treatable. Annu Rev Med 2020; 71: 203-19 9 Bonderman D et al.: Diagnosis and treatment of cardiac amyloidosis: an interdisciplinary consensus statement. Wien Klin Wochenschr 2020; 132(23-24): 742-61 10 McDonagh TA et al.: 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2021; 42(36): 3599-726 11 Garcia-Pavia P et al.: Diagnosis and treatment of cardiac amyloidosis: A position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 2021; 42(16): 1554-68 12 Kastritis E et al.: Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med 2021; 385(1): 46-58 13 Maurer MS et al.: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379(11): 1007-16
Das könnte Sie auch interessieren:

Mechanische Kreislaufunterstützung im Infarkt-bedingten kardiogenen Schock
Der Infarkt-bedingte kardiogene Schock (AMI-CS) ist trotz der enormen Fortschritte in der interventionellen Versorgung des akuten Myokardinfarktes in den vergangenen Jahrzehnten ...
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...