HCM-Therapie in der klinischen Praxis
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
HCM wird mit Betablockern oder Kalziumantagonisten behandelt, bringt dies keinen Erfolg, blieb bis vor Kurzem als einzige Alternative die invasive Septumreduktion (SRT). Nun steht mit dem Myosin-Inhibitor Mavacamten eine neue medikamentöse Option zur Verfügung, die kausal in den Pathomechanismus der HCM eingreift.
Therapie der HCM
Die HCM ist definiert als nachlastunabhängige Hypertrophie des linken Ventrikels(LV) ≥15mm ohne kardiale Grunderkrankung wie Hypertonie oder Aortenstenose. „Leitsymptome sind Belastungs- oder Ruhedyspnoe, Angina pectoris, Palpitationen und Schwindel, wobei eine HCM häufig asymptomatisch ist und zufällig im Rahmen einer Gesundenuntersuchung entdeckt wird“, so Univ.-Prof. Dr. Marc-Michael Zaruba, Universitätsklinik für Innere Medizin III, Innsbruck. Die HCM ist mit einer Prävalenz von 1:500 relativ häufig. Die Prognose hängt von der Ätiologie ab. So weist die Amyloid-Kardiopathie mit einem 10-Jahres-Überleben von weniger als 40% eine hohe Mortalität auf.1 Auch der Phänotyp beeinflusst die Prognose, wobei die obstruktive HCM (HOCM) ein höheres Progressionsrisiko (7,4%) für eine fortgeschrittene Herzinsuffizienz (NYHA III/IV; NYHA: New York Heart Association) hat. Bei nicht obstruktiver HCM ist das Risiko höher, wenn unter Belastung eine Obstruktion induziert werden kann. „Je höher der Druckgradient, desto schlechter ist die Prognose“, so Zaruba. Die klinische Evaluation ist also wichtig (Abb. 1).2
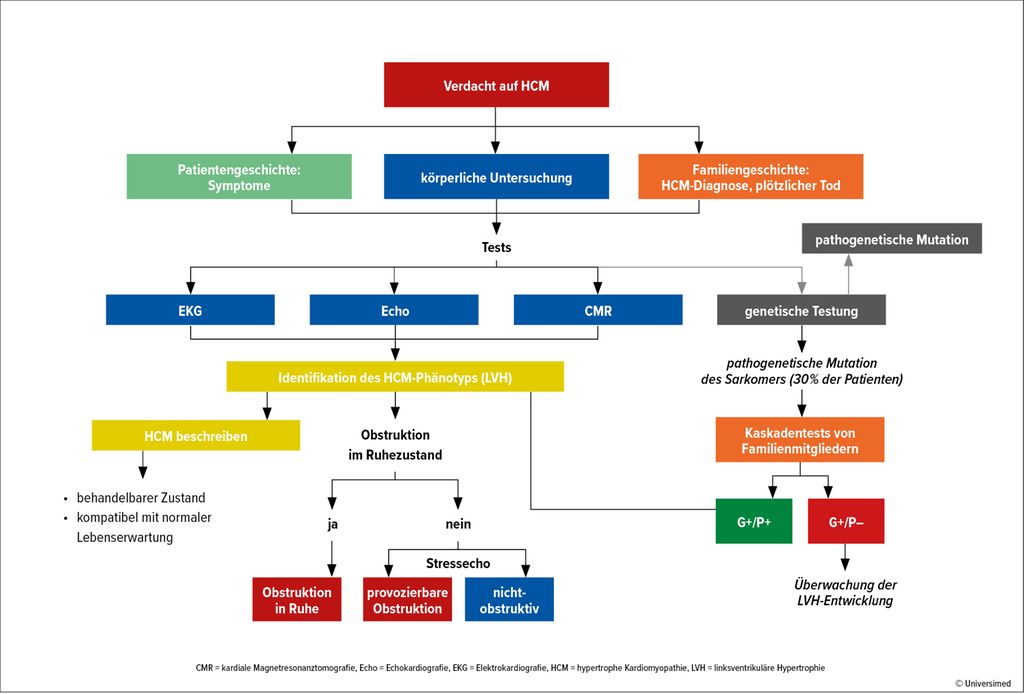
Abb. 1: Klinische Evaluation bei Verdacht auf hypertrophe Kardiomyopathie (nach Maron BJ et al. 2022)2
Die Therapiempfehlungen hängen vom Phänotyp und der Symptomatik ab.Substanzen erster Wahl sind Betablocker sowie die Kalziumantagonisten Verapamil und Diltiazem. Kommt es unter Therapie zur Progression, sind bei HOCM die chirurgische Myektomie oder die alkoholische Septumablation indiziert. Bei nicht obstruktiver HCM kann ein implantierbarer Defibrillator (ICD) indiziert sein oder eine Herztransplantation.
Mit Mavacamten, einem allosterischen, selektiven und reversiblen Inhibitor des kardialen Myosins, steht in der HCM-Therapie nun ein spezifisch wirksames, kausal in den Pathomechanismus eingreifendes Medikament zur Verfügung. Mavacamten setzt bei der für HCM typischen Hyperkontraktilität des Herzmuskels an. Diese wird durch die reversible und spezifische Inhibition von Myosin reduziert. Myosin wird in einem inaktiven, energiesparenden und superrelaxierten Zustand stabilisiert, was zur Verringerung der LV-Obstruktion und zur Verbesserung des kardialen Füllungsdrucks führt.3,4 Die Zulassung von Mavacamten beruht auf den Phase-III-Studien EXPLORER-HCMund VALOR-HCM.5,6 EXPLORER-HCM zeigte, dass Mavacamten bei HOCM-Patienten im Vergleich zu Placebo die maximale Sauerstoffaufnahme und die NYHA-Klasse verbessert. In Ruhe und unter Belastung wurden relevante Reduktionen des Ausflusstraktgradienten ohne signifikante Einschränkungen der Pumpfunktion beobachtet.
Weniger interventionelle Therapien
In VALOR-HCM wurde mit dem Bedarf nach chirurgischer SRT ein harter klinischer Endpunkt gewählt. Eingeschlossen wurden Patienten mit HOCM, bei denen trotz maximal verträglicher medikamentöser Therapie eine SRT indiziert war. Primärer Endpunkt war die Kombination aus Patientenentscheidung für eine SRT sowie deren Indikation gemäß AHA-/ESC-Leitlinie.7 Nach 16 Wochen erfüllten nur noch 17,9% der Verum-, aber noch 76,8% der Placebopatienten diese Anforderung. In der Folge erhielten alle Patienten Mavacamten. Nach weiteren 16 Wochen sank auch in der ehemaligen Placebogruppe der Bedarf nach SRT signifikant und deutlich auf 14%. Bei den kontinuierlich mit Mavacamten behandelten Patienten erfüllten nach insgesamt 32 Wochen Therapie nur noch 11% der Patienten die Kriterien für eine SRT oder bevorzugten den Eingriff. Die NYHA-Klasse verbesserte sich, es kam zur Reduktion des Druckgradienten im LV-Ausflusstrakt sowohl in Ruhe als auch beim Valsalva-Manöver sowie von NT-proBNP und Troponin.
Mavacamten in der klinischen Praxis
Wie der Einsatz von Mavacamten im klinischen Alltag aussehen kann, erläutert Dr. Moritz Messner, Universitätsklinik für Innere Medizin III, Innsbruck, am Beispiel eines Patienten, der vor mehr als 10 Jahren mäßig symptomatisch mit einem NT-proBNP von 280ng/l an seinem Zentrum vorstellig wurde. Im EKG fanden sich hohe Amplituden als Indiz für eine LV-Hypertrophie, die echokardiografisch bestätigt wurde. Es zeigte sich eine Flussbeschleunigung im Ausflusstrakt. Unter Belastung traten ein Druckgradient mit einem spätsystolischen Maximum bis 158mmHg, eine Mitralklappeninsuffizienz (MI) und ein SAM-Phänomen („systolic anterior motion“) auf.
In der Familie des Patienten waren keine Fälle von plötzlichem Herztod (SCD) bekannt. Es gab keine Synkopen und im Langzeit-EKG keine Hinweise auf ventrikuläre Tachykardien. Die Belastbarkeit in der Ergometrie war gut. So ergab der SCD-Risiko-Score zur Risikostratifikation ein geringes SCD-Risiko, womit keine Indikation zur ICD-Implantation bestand.
Gemäß Leitlinien wurde eine Betablockertherapie begonnen und mit dem Patienten die Option einer chirurgischen SRT bei Krankheitsprogression besprochen. Kontrollen wurden in Sechsmonatsintervallen vereinbart.Die genetische Testung ergab eine MYBPC3-Mutation unklarer Signifikanz, womit keine prädiktive Genetik bei Verwandten indiziert ist.
2023 kam der Patient indie Ambulanz und klagte über abnehmende Leistungsfähigkeit. In der Auskultation war ein Systolikum zu hören, Kongestionszeichen gab es nicht. Die Echokardiografie zeigte eine Septumdicke von 19–20mm, einen maximalen Ruhegradienten von 80mmHg, der bei Belastung auf 150mmHg anstieg, sowie ein SAM-Phänomen und eine MI. Das NT-proBNP betrug 700ng/l. Zunächst wurde an eine SRT gedacht, jedoch wegen der erfolgten EU-Zulassung von Mavacamten und dem Update der HCM-Leitlinien der europäischen Gesellschaft für Kardiologie (ESC), fiel die Entscheidung auf Mavacamten. Gemäß der Mavacamten Fachinformation waren die Voraussetzungen dafür (NYHA II/III und LVEF >55%) erfüllt. Vor Therapiebeginn wurde eine CYP2C19-Genotypisierung durchgeführt, da bei langsamen Metabolisierern dreimal höhere Plasmaspiegel auftreten können. Eine gemeinsame Einnahme von Mavacamten und CYP2C19-Inhibitoren (z.B. Omeprazol oder Esomeprazol) war zu vermeiden. Der Patient war ein intermediärer Metabolisierer und wurde somit im Oktober 2023 auf eine Startdosis von 5mg Mavacamten eingestellt. Die Titration erfolgt auf Basis von Kontrollen nach jeweils vier Wochen, wobei der linksventrikuläre Auswurftrakt(LVOT)-Gradient unter Belastung gemessen wird. Solange dieser laut Fachinformation ≥20mmHg bleibt, wird die Startdosis von 5mg beibehalten. Fällt er, muss die Dosis reduziert oder die Behandlung unterbrochen werden. Nach 12 Wochen kann eine Dosissteigerung angedacht werden. Die Therapie muss für vier Wochen pausiert werden, wenn die linksventrikuläre Ejektionsfraktion (LVEF) unter 50% abfällt.
Der Patient zeigte nach 12 Wochen unter 5mg Mavacamten keinen Druckgradienten in Ruhe und unter Belastung einen von 73mmHg. Das NT-proBNP lag knapp über dem Normalbereich. Nun ist eine Dosissteigerung geplant. Mavacamten kann bis zu 15mg aufdosiert werden. Nach erfolgter Abklärung erhielt der Patient die Freigabe für vorsichtigen Ausdauersport und kann wieder Skitouren gehen.
Mavacamten (Camzyos®) ist in Österreich in der Indikation HOCM verfügbar. Laut den aktuellen ESC-Leitlinien besteht für Mavacamten eine Klasse-IIa-Empfehlung für HOCM-Patienten, die mit Betablockern und Kalziumantagonisten symptomatisch bleiben.9
Quelle:
Satellitensymposium Bristol-Myers Squibb Ges.m.b.H. „Myosin-Inhibierung – die erste kausale Therapie in der HOCM“, 7.3.2024, Kardiologie-Kongress Innsbruck
Literatur:
1 Kocher F et al.: ESC Heart Fail 2020; 7(6): 3919-28 2 Maron BJ et al.: J Am Coll Cardiol 2022; 79(4): 390-414 3 Rohde JA et al.: Proc Natl Acad Sci USA 2018;115(32); E7486-94 4 Heitner SB et al.: Ann Intern Med 2019; 170(11): 741-8 5 Olivotto I et al.: Lancet 2020; 396(10253): 759-69 6 Desay MY et al.: J Am Coll Cardiol 2022; 80(2): 95-108 7 Elliott PM et al.: Eur Heart J 2014; 35(39): 2733-79 8 Fachinformation Camzyos®: Stand August 2023 9 Arbelo E et al.: Eur Heart J 2023; 44(37): 3503-626
Entgeltliche Einschaltung
Mit freundlicher Unterstützung durch Bristol-Myers Squibb Ges. m. b. H.
Fachkurzinformation | 3500-AT-2400021, 04/2024