
Hypertrophe Kardiomyopathie: häufiger als gedacht
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die hypertrophe Kardiomyopathie (HCM) ist eine häufige, jedoch unterdiagnostizierte Erkrankung. Sie ist meist genetisch bedingt und macht sich oft erst dann bemerkbar, wenn Symptome einer Herzinsuffizienz auftreten. Plötzlicher Herztod und durch Vorhofflimmern verursachte Thromboembolien sind die häufigsten Komplikationen.1,2
Die ersten Anzeichen für eine HCM erkennen
Potenzielle HCM-Indikatoren:7,8
-
Symptome wie Dyspnoe, Angina pectoris und körperliche Belastungsintoleranz
-
Auffälliges EKG
-
Systolikum
-
Positive Familienanamnese
Definiert ist die HCM als eine linksventrikuläre Hypertrophie (LVH), die nicht durch eine andere kardiale, systemische oder metabolische Erkrankung erklärt werden kann, erläuterte Dr. Pablo Garcia-Pavia, Department of Cardiology of Hospital Universitario Puerta de Hierro, Madrid, am Jahreskongress der European Society of Cardiology (ESC).*,1
Die HCM ist eine häufige Erkrankung: Man geht von einer weltweiten Prävalenz von 1:500, aktuellen Daten zufolge sogar von 1:200 aus.3 In Österreich wären demzufolge etwa 18000 bis 45000 Personen betroffen. Zum Vergleich: Das größte Fußballstadion in Österreich, das Ernst-Happel-Stadion in Wien, fasst ca. 50000 Personen. Viele Patienten mit HCM bleiben jedoch undiagnostiziert.4 Einen Grund dafür sieht der Experte darin, dass viele der Patienten keine klinisch signifikanten Symptome haben und zufällig – in der Regel mit einer leichten Krankheitsausprägung – identifiziert werden.1
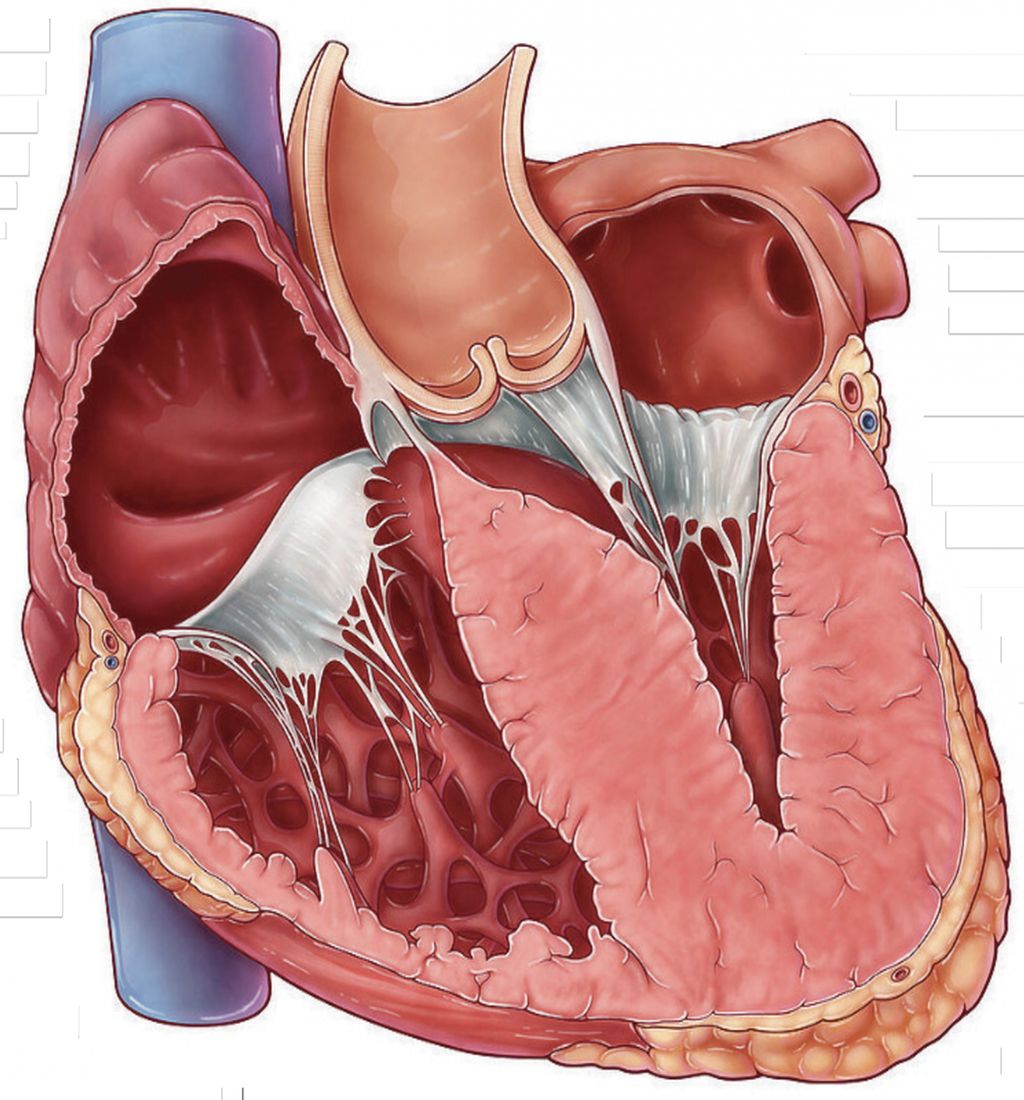
Abb. 1: Schemazeichnung hypertrophe Kardiomyopathie
Das frühe Erkennen der Krankheit ist wichtig, da HCM das Risiko für kardiovaskuläre Ereignisse erhöht: So haben Betroffene eine drei- bis vierfach erhöhte Gesamtmortalität im Vergleich zur Allgemeinbevölkerung, ein doppelt so hohes Risiko für eine Herzinsuffizienz und eine dreifach erhöhte Rate an Vorhofflimmern.2 Beim Großteil der Patienten wird die HCM durch einen Gendefekt im Sarkomer verursacht. Dadurch kommt es zu einer Zunahme von Myosin-Aktin-Querbrückenverbindungen. Die Dysfunktion der Sarkomere, vor allem die Hyperkontraktilität und die gestörte Relaxation, führt zu Herzmuskelhypertrophie, häufig begleitet von einer Fibrose (Abb. 1).5,6
Dem plötzlichen Herztod vorbeugen
Ein wichtiger Aspekt im Management der Patienten sind die Abschätzung und die Prävention des plötzlichen Herztods.7 Hierfür gibt es in den USA und in Europa zwei unterschiedliche Herangehensweisen, wie der Experte erläuterte. In Europa sind die ESC-Guidelines aus dem Jahr 2014 gebräuchlich, die einen Kalkulator zur Abschätzung des plötzlichen Herztods verwenden.7 Anhand des berechneten Risikos diskutiert man mit dem Patienten, ob ein implantierbarer Cardioverter-Defibrillator (ICD) nötig ist oder nicht.
Die US-amerikanischen HCM-Guidelines aus dem Jahr 2020 basieren dagegen auf individuellen Risikofaktoren für den plötzlichen Herztod. Bereits bei Vorliegen eines Risikofaktors, etwa Vorhofflimmern oder eine Auswurffraktion ≤50%, kann die Implantation eines ICD erfolgen.8
Die Prävention kardioembolischer Komplikationen aufgrund von Vorhofflimmern ist ein weiterer wichtiger Aspekt bei HCM. Einer Metaanalyse zufolge liegt die Prävalenz von Vorhofflimmern bei HCM-Patienten bei etwa 22% und die damit in Zusammenhang stehenden thromboembolischen Komplikationen liegen bei 27% – mit einer Inzidenz von 3,75 pro 100 Patienten pro Jahr.9 Diese Zahlen sind bemerkenswert, so der Experte,weil es sich um eine Population von Menschen ohne die traditionellen kardiovaskulären Risikofaktoren handelt.*
Obstruktion verursacht Herzinsuffizienzsymptome
Die LVH beeinträchtigt die ventrikuläre Füllung und ist häufig mit einer diastolischen Dysfunktion assoziiert.10 Die HCM kann daher zu Herzinsuffizienz und den damit verbundenen Symptomen führen. Bei ca. 70% der HCM-Patienten liegt gleichzeitig eine Obstruktion des linksventrikulären Ausflusstrakts (LVOT) vor. Diese wird jedoch häufig erst unter Belastung symptomatisch.10,11 Dr. Garcia-Pavia wies besonders darauf hin, dass das Vorliegen einer Obstruktion bei HCM ein unabhängiger Risikofaktor für kardiovaskuläre Ereignisse ist.12
Reine Vasodilatatoren und Diuretika vermeiden
Die therapeutischen Optionen für die symptomatische obstruktive HCM sind limitiert. Es stehen medikamentöse und interventionelle Maßnahmen zur Verfügung. Die Pharmakotherapie umfasst nichtvasodilatierende Betablocker, Kalziumkanalblocker (vor allem Verapamil und Diltiazem) und Disopyramid. Substanzen, welche die Ausflusstraktobstruktion weiter negativ beeinflussen wie reine Vasodilatatoren (ACE-Hemmer, Sartane, Dihydropyridin) sowie Diuretika in hoher Dosis sind zu vermeiden.8 Als interventionelle Maßnahmen kommen die septale Myektomie oder die transkoronare Ablation der Septumhypertrophie (TASH) im weiteren Verlauf in Betracht.13
Die Echokardiografie ist diagnostisch wegweisend
Für die Diagnostik der hypertrophen Kardiomyopathie ist die Bildgebung wegweisend. An erster Stelle steht dabei die Echokardiografie.
Eine HCM klinisch zu erkennen ist nicht so einfach, erklärte Professor Dr. Jeroen J. Bax, Department of Cardiology, Leiden University Medical Center. Die Symptome sind unspezifisch und überlappen mit denen anderer pulmonaler oder kardiovaskulärer Erkrankungen.14 Nach den AHA/ACC-Guidelines von 2020 ist die kardiale Bildgebung in Ruhe und unter Belastung der Standard, um die Diagnose HCM zu stellen.8 Die Echokardiografie ist die erste Wahl, um die kardiale Morphologie darzustellen und eine linksventrikuläre Hypertrophie bzw. eine LVOT-Obstruktion zu entdecken.8,15 Dabei ist die Dicke der linksventrikulären Wand der relevante Parameter für die Diagnose. Nach den Guidelines7,8 muss die maximale enddiastolische Wanddicke an irgendeiner Stelle im linken Ventrikel ≥15mm betragen. Bei Familienangehörigen von Patienten mit HCM oder einer genetischen Mutation sind bereits 13mm diagnostisch wegweisend. Bei Kindern gilt ein adjustierter Z-Score von ≥2 Standardabweichungen vom Mittelwert.7,8 Alle linksventrikulären Segmente können von der Verdickung betroffen sein.7
Für die Prognose ist die Wanddicke ebenfalls von Bedeutung, denn es gibt eine lineare Beziehung zwischen ihr und dem plötzlichen Herztod.16 Das höchste Risiko besteht bei einem Wert von ≥30mm.16 Die LVOT-Obstruktion resultiert aus dem mitralseptalen Kontakt, verursacht durch septale Hypertrophie und/oder eine Vorwölbung des vorderen Mitralklappensegels gegen das Septum (SAM, „systolic anterior motion“).8
Da die LVOT-Obstruktion dynamisch sein kann und eine obstruktive HCM in Ruhe nicht immer vorhanden ist, gilt die Empfehlung, stets ein Valsalva-Manöver oder ein Stress-Echo durchzuführen, wenn der Ruhe-Ausflusstraktgradient <50mmHg beträgt, so Dr. Bax.8
In Zweifelsfällen kann das MRI helfen, erklärte der Experte. Damit ist vor allem eine myokardiale Fibrose gut zu erkennen. Die Guidelines empfehlen (Klasse I, Level B) ein kardiales MRT bei Patienten mit vermuteter HCM, die sich in der Echokardiografie nicht adäquat darstellen lässt.8 Die Fibrose ist ein Marker für Krankheitsprogression und ein erhöhtes Risiko für Herzinsuffizienz und plötzlichen Herztod.17 Patienten mit Fibrose sollten daher engmaschig monitorisiert werden.17
Quelle:
*Satellitensymposium „Identifying obstructive HCM: expert perspectives“ von Bristol-Myers Squibb. ESC-Jahreskongress 2022, Barcelona, 26. August 2022
Literatur:
1 Maron BJ et al.: Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med 2018; 379(7): 655-68 2 Ho CY et al.: Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation 2018; 138(14): 1387-98 3 Maron BJ et al.: Global burden of hypertrophic cardiomyopathy. JACC Heart Fail 2018; 6(5): 376-8 4 Maron MS et al.: Occurrence of clinically diagnosed hypertrophic cardiomyopathy in the United States. Am J Cardiol 2016; 117(10): 1651-4 5 Spudich JA et al.: Three perspectives on the molecular basis of hypercontractility caused bei hypertrophic cardiomyopathy mutations. Pflugers Arch 2019; 471(5): 701-17 6 Kamisago M et al.: Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med 2000; 343(23): 1688-96 7 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39): 2733-79 8 Ommen SR et al.: 2020 AHA/ACC Guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy. Circulation 2020; 142(25): e558-e631 9 Guttmann OP et al.: Atrial fibrillation and thromboembolism in patients withhypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6): 465-72 10 Marian AJ, Braunwald E: Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 2017; 121(7): 749-70 11 Maron MS et al.: Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21): 2232-9 12 Lu DY et al.: Clinical outcomes in patients with nonobstructive, labile, and obstructive hypertrophic cardiomyopathy. J Am Heart Assoc 2018; 7(5): e006657 13 Ommen SR et al.: Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3): 470-6 14 Arulian E et al.: Misconceptions and facts about hypertrophic cardiomyopathy. Am J Med 2016; 129(2): 148-52 15 Nagueh SF et al.: American Society of Echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: endorsed by the American Society of Nuclear Cardiology, Society for Cardiovascular Magnetic Resonance, and Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr 2011; 24(5): 473-98 16 Gersh MB et al.: 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy. J Am Coll Cardiol 2011; 124(24): 2761-96 17 O’Hanlon R et al.: Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11): 867-74
Entgeltliche Einschaltung
Mit freundlicher Unterstützung durch Bristol-Myers Squibb Ges.m.b.H.
CV-AT-2300018, 05/23