
Welche Lipoproteine sind ursächlich für Atherosklerose und Co.?
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Im klinischen Alltag und in Diagnose-Scores werden zahlreiche Faktoren zur Bestimmung des Risikos für Herzinfarkt, Diabetes mellitus oder Schlaganfall herangezogen. Auch die Lipoproteine finden hier Anwendung. Doch was wissen wir darüber, ob zwischen Faktor und Erkrankung ein Kausalzusammenhang besteht – oder wir nicht vielleicht nur einen Störfaktor messen, der an der Pathogenese unbeteiligt ist? Den aktuellen Stand der Evidenz legte Prof. Dr. med. Arnold von Eckardstein in seinem Vortrag beim AGLA-Kurs «Klinische Lipidologie» in Zürich dar.
Keypoints
-
Die kausale Rolle von LDL-C bei der Atherosklerose ist wissenschaftlich gut belegt.
-
Für die Triglyzeride zeigen genetische Studien einen Zusammenhang mit dem Entstehen einer Atherosklerose; Resultate aus Interventionsstudien sind aber inkonsistent.
-
Für das Lipoprotein(a) liefern genetische Studien und Tiermodelle starke Hinweise auf eine Begünstigung der Atherosklerose.
-
HDL-C ist kein kausaler Risikofaktor, sodass HDL-C erhöhende Therapien nicht indiziert sind, zumal ein hohes HDL-C mit erhöhter Mortalität assoziiert ist.
Eine Beobachtung in einer epidemiologischen Studie beweise keinen Kausalzusammenhang – auch wenn man sie bei tausenden von Menschen mache, sagte Prof. Dr. med. Arnold von Eckardstein, Institut für Klinische Chemie des Universitätsspitals Zürich, und erinnerte an die Regeln zur Beurteilung von Kausalität. Der Goldstandard zur Überprüfung von Kausalität ist die randomisierte, kontrollierte, verblindete Interventionsstudie (RCT). Hier werden Confounder, also Störfaktoren, die das Ergebnis beeinflussen können, durch Randomisierung gleichmässig auf die Studiengruppen verteilt und somit weitestgehend ausgeschaltet. Auch das in Beobachtungsstudien mögliche Phänomen der reversen Kausalität – dass also nicht die Veränderung eines Faktors die Erkrankung verursacht, sondern die Erkrankung den Faktor verändert – ist durch das Studiendesign bei RCTs ausgeschaltet.
«Entdeckt man in Beobachtungsstudien also einen Risikofaktor für eine Erkrankung, ist es für die Entwicklung medikamentöser Therapien, die diesen Faktor günstig beeinflussen sollen, essenziell, vorab zu sichern, dass auch wirklich ein kausaler Zusammenhang besteht. Während hier lange Zeit die biologische Plausibilität in Versuchen mit Zellkulturen und Tiermodellen zu sichern versucht wurde, setzt sich heute immer mehr die Strategie der Mendel’schen Randomisierung durch. Das Grundprinzip liegt hier in der durch Vererbung zufälligen Verteilung elterlicher Allele, die für einen bestimmten Risiko- oder protektiven Faktor codieren. Mutationen in diesen Allelen können so aus genetischer Sicht kausal mit einem erhöhten oder einem erniedrigten Risiko für eine Erkrankung in Verbindung gebracht werden»,1 erklärte der Experte.
Als Beispiel führte von Eckardstein einige Marker an, die in Beobachtungsstudien mit einem erhöhten Herzinfarktisiko assoziiert waren. «Erst die Untersuchung der genetischen Varianten dieser Faktoren hinsichtlich ihres Einflusses auf das Herzinfarktrisiko haben gezeigt, ob der Effekt wirklich kausal ist oder nicht. Eine genetische Variante führt sowohl zu einer Veränderung des durch sie codierten Faktors als auch zur Veränderung des Erkrankungsrisikos. Für LDL-Cholesterin (LDL-C), Triglyzeride, Lipoprotein(a) und sogar den Body Mass Index indes konnten so genetische Kausalitätsbezüge zur Atherosklerose gezeigt werden. Für HDL-Cholesterin (HDL-C) dagegen hat die Überprüfung in Mendel’schen Randomisierungsstudien keinen genetischen Kausalzusammenhang ergeben; ebensowenig für C-reaktives Protein (CRP), Homocystein, Fibrinogen oder die Lipoprotein-assoziierte Phospholipase A2 (Lp-PLA2). Die Genetik hilft uns also dabei, die Frage nach dem Kausalzusammenhang von Faktoren und Erkrankungen zu beantworten – und letztlich bei der Klärung, ob ein bestimmter Faktor überhaupt einen sinnvollen Ansatzpunkt für eine Therapie darstellt.»2
Wie ist es aber nun im Einzelnen um die Evidenz für oder gegen eine kausale Beziehung zwischen verschiedenen Lipoproteinen und dem Risiko für Atherosklerose sowie weitere Erkrankungen bestellt? Und welche Erkenntnisse zu Erkrankungsrisiken durch Korrekturen von Risikofaktoren können wir durch genetische Studien gewinnen?
LDL-Cholesterin – gut untersucht, meist schuldig
LDL-Cholesterin und Herzinfarktrisiko
LDL-C wie auch NonHDL-C und ApoB zeigen in Beobachtungsstudien eine positive Assoziation mit dem Herzinfarktrisiko – je höher der LDL-C-Spiegel, desto höher das Risiko. Dieses konnte in RCTs mit LDL-Senkern (Statine, PCSK9-Hemmer, Ezetimib, Bempedoinsäure) gesenkt werden. Auch in genetischen Studien sah man, dass die Genvarianten, die das LDL-C erhöhen oder senken, eine sehr enge Beziehung zum Herzinfarktrisiko aufwiesen – bedingt durch die lange Beobachtungsdauer von im Median 52 Jahren sogar eine viel stärkere Beziehung als in Beobachtungsstudien und RCTs mit 12 bzw. 5 Jahren Nachbeobachtung. «Auch in Tiermodellen, die ein wichtiges Kriterium bei der Plausibilitätsüberprüfung sind, sieht man, dass es nur zur Atherosklerose kommt, wenn man eine Hypercholesterinämie induziert»,3 ergänzte von Eckardstein.
Ein erhöhtes LDL-C ist also eine notwendige, nicht aber eine hinreichende Voraussetzung für das Entstehen der Atherosklerose. Das ist im LDL-Katabolismus begründet: Im Normalfall wird LDL zu 95% über LDL-Rezeptoren der Leber aufgenommen und entfernt, und nur ein ganz kleiner Teil wird über andere Zellen, vor allem Makrophagen, über Scavenger-Rezeptoren entsorgt. Ist der Weg über die Leber gestört, beispielsweise durch Mutationen im LDL-Rezeptor-Gen wie bei der familiären Hypercholesterinämie oder durch eine ernährungsbedingte LDL-Rezeptor-Herabregulierung, kommt es zur Hypercholesterinämie und damit zur vermehrten Aufnahme von enzymatisch oder oxidativ modifiziertem LDL in die Makrophagen. «Da die für die Aufnahme verantwortlichen Scavenger-Rezeptoren im Gegensatz zum LDL-Rezeptor nie herunterreguliert werden, also quasi immer ‹an› sind, nehmen die Makrophagen nahezu unlimitiert LDL auf», sagte von Eckardstein. Auf diese Weise funktioniert auch die Atheroskleroseentstehung (Abb. 1): LDL aktiviert – gemeinsam mit Faktoren wie erhöhtem Blutdruck oder Hyperglykämie – das Endothel, was zur LDL-Aufnahme in die Arterienwand (1) und zur Einwanderung von Monozyten in die Intima (2) führt. Dort wandeln sie sich in Makrophagen um (3). Über die Scavenger-Rezeptoren nehmen diese oxidiertes LDL auf (4) und werden zu Schaumzellen (6), die eine massive entzündliche Reaktion mit Produktion von Zytokinen und Wachstumsfaktoren hervorrufen. Das führt zur weiteren Einwanderung von Leukozyten (5) sowie zur Proliferation der glatten Muskelzellen der Media, die in die Intima einwandern (7, 8) und sich dort in bindegewebsartige Zellen umwandeln. Durch Ausbilden einer fibrösen Kappe versuchen sie, den Entzündungsherd abzuschirmen. «Das ist das, was wir dann in der Bildgebung als Plaque oder fibröse Kappe sehen, zunehmend mit Verkalkungen», erläuterte von Eckardstein. «Das allein macht aber noch keinen Herzinfarkt. Erst wenn die Plaque durch Proteasen aus den Makrophagen aufgelöst wird und schliesslich einreisst, kommt es durch den Kontakt des Blutes mit dem Bindegewebe zur Aktivierung von Thrombozyten und Gewebefaktor und somit zur Ausbildung eines Thrombus. Das führt dann zum Gefässverschluss und damit klinisch zum Infarkt. Da LDL direkt oder indirekt massgeblich an allen Schritten der Atherogenese beteiligt ist, wird es auch zu Recht als Haupttreiber der Atherogenese bezeichnet.4 Seine Kausalität kann nahezu als bewiesen angesehen werden, da RCTs, Mendel’sche Randomisierung und Tiermodelle zu konsistenten Ergebnissen geführt haben und die Effektivität einer medikamentösen LDL-Senkung durchgehend bestätigt werden konnte.»5
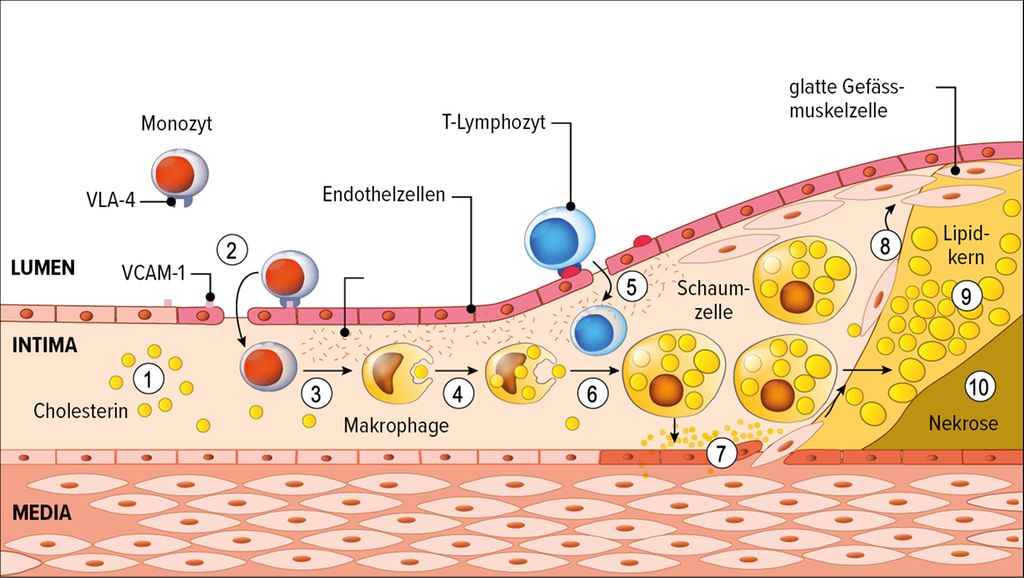
Abb. 1: Entstehung der Atherosklerose: LDL-Cholesterin (1) als Auslöser der Prozesse in der Gefässintima
LDL-Cholesterin und Diabetesrisiko
Auch eine ganze Reihe anderer Erkrankungen steht im Zusammenhang mit LDL-C. Beim Diabetes mellitus wurde eine inverse Beziehung beobachtet, niedrige LDL-Werte erhöhen also das Diabetesrisiko. RCTs mit LDL-Senkung durch Statine bestätigten diese Beobachtung, die Diabetesinzidenz stieg gegenüber Placebo um 9%, bei intensiver LDL-Senkung vs. moderate um 12% an. Für die anderen LDL-Senker, Ezetimib, PCSK9i und Bempedoinsäure, zeigte sich dieser Zusammenhang nicht.6 «Eine Erklärung dafür könnte sein, dass Ezetimib und PCSK9i immer on top zu den Statinen dazugegeben worden sind – d.h., diejenigen mit Prädiabetes hatten ihn möglicherweise einfach schon unter der Statintherapie manifestiert. Eine andere Erklärung liegt im Pathomechanismus der medikamentösen Therapien begründet: Statine regeln den LDL-Rezeptor hoch, und zwar nicht nur in den Leberzellen, sondern in allen Zellen des Körpers. So auch in den Betazellen des Pankreas, in denen durch die vermehrte LDL-Aufnahme der Cholesteringehalt ansteigt, was die Insulinsekretion stört. Beim Gesunden sollte das keinen grösseren klinischen Effekt haben, aber bei Menschen mit metabolischem Syndrom und bereits erhöhter Insulinresistenz ist das möglicherweise für die Entstehung eines manifesten Diabetes ausreichend», sagte von Eckardstein. «Die PCSK9-Inhibitoren wirken dagegen nur an den Leberzellen, indirekt auch Ezetimib via Hemmung der Cholsterinaufnahme im Darm – die Betazellen des Pankreas werden nicht beeinflusst.7 Bei der Bempedoinsäure könnte der fehlende Einfluss auf das Diabetesrisiko eventuell daran liegen, dass es sich um ein Prodrug handelt, das erst enzymatisch aktiviert werden muss, und dass die Betazellen nicht über dieses Enzym verfügen»
Dass der Zusammenhang zwischen LDL und Diabetes kausal ist, legen die Ergebnisse aus genetischen Studien und Tiermodellen nahe, und das dürfe laut dem Experten auch nicht geleugnet werden. Trotzdem müsse man immer das Nutzen-Risiko-Verhältnis bedenken: In einer genetischen Studie konnte gezeigt werden, dass eine experimentelle Erhöhung des LDL-Wertes um eine Standardabweichung das Diabetesrisiko um 10% erniedrigt, aber gleichzeitig das kardiovaskuläre Risiko um 60% erhöht. Damit überwiegt für von Eckardstein eindeutig der Nutzen der LDL-Senkung in Bezug auf die Atherosklerose und ihre Folgeerkrankungen.8
LDL-Cholesterin und Schlaganfallrisiko
Ebenfalls eine inverse Beziehung wurde in Bezug auf den hämorrhagischen Schlaganfall beobachtet. Auch hier geben Interventions- und genetische Studien starke Kausalitätshinweise. «Die Studiendaten zeigen eindrücklich, dass mit zunehmendem LDL-C das Risiko für den ischämischen Schlaganfall steigt, das für den hämorrhagischen aber sinkt. Das klingt zwar erstmal nicht gut, sollte aber nicht gleich zum Grund gegen eine medikamentöse LDL-Senkung werden. Denn bedenkt man, wie selten der hämorrhagische Schlaganfall ist, übersteigt der Nutzen der LDL-Senkung und damit der Verhinderung von ischämischen Schlaganfällen bei Weitem das Risiko für den hämorrhagischen Schlaganfall unter niedrigeren LDL-Werten»,9 gab von Eckardstein zu bedenken.
LDL-Cholesterin und Risiko für Fettleber oder Gallensteine
Hinsichtlich des klinischen Endpunktes der Fettleber wurde epidemiologisch kein erhöhtes Risiko in Abhängigkeit vom LDL-Spiegel beobachtet, wohl aber unter Intervention mit bestimmten Medikamenten (Mipomersen, Lomatipid). In der Folge zeigten Mendel’sche Studien dies als genabhängig. Nur Varianten, welche die Produktion Apo-B-haltiger Lipoproteine vermindern, erhöhten das Risiko einer Fettleber, nicht aber Varianten, welche den Katabolismus von Apo-B-haltigen Lipoproteinen steigern. In RCT erhöhte die Behandlung mit Ezetimib das Risiko einer Cholelithiasis. Genetische Varianten, welche wie Ezetimib die Cholesterinresorption im Darm hemmen, sind ebenfalls mit einem erhöhten Risiko für Gallensteine assoziiert.
LDL-Cholesterin und Krebsrisiko
Bei Krebserkrankungen zeigte sich in Beobachtungsstudien ein erhöhtes Risiko für Menschen mit niedrigen LDL-Spiegeln, in Interventions- und genetischen Studien konnte aber kein Signal für einen kausalen Zusammenhang gesehen werden, ebensowenig in Tiermodellen. «Man nimmt an, dass das niedrige LDL nicht die Ursache für die Krebserkrankung, sondern eine Folge des Krebses ist», so von Eckardsteins Interpretation.
LDL-Cholesterin und Demenzrisiko / LDL-Cholesterin und Mortalität
Für die Demenz gibt es keinerlei Hinweise für einen Zusammenhang mit den LDL-Werten, während sich bei der Mortalität eine parabolische (J-förmige) Assoziation beobachten liess: Eine hohe Mortalität wurde sowohl bei niedrigen als auch bei hohen LDL-Werten gesehen. Hier weisen die Ergebnisse aus Interventions- und genetischen Studien nur für die hohen LDL-Werte, nicht aber für die niedrigen, auf einen kausalen Zusammenhang hin und bestätigten für diesen Bereich den protektiven Effekt einer LDL-Senkung.
Triglyzeride – gesicherter Auslöser für Atherosklerose und Co.?
«Bei den Triglyzeriden wurden in epidemiologischen Beobachtungsstudien ebenfalls Beziehungen zu Herzinfarkt, Diabetes, Fettleber und Mortalität gesehen, ebenso zur akuten Pankreatitis», erklärte von Eckardstein. «Doch wenn es um die Kausalität geht, ist unser Wissensstand noch lange nicht so weit wie beim LDL-C.» Bei der Atherosklerose sprechen die Ergebnisse aus den genetischen Studien zwar für Kausalität10, die Interventionsstudien zur Triglyzeridsenkung – sei es mit Fibraten, Omega-3-Fettsäuren oder Niacin – lieferten jedoch sehr inkonsistente Ergebnisse. Im Tiermodell finden wir eine mögliche Erklärung dafür, haben wir doch gesehen, dass es sehr vom Gen abhängt, das man verändert, ob ein Effekt auf die Atheroskleroseentstehung gesehen wird oder nicht.» Dieses Konzept wird derzeit mit Medikamenten geprüft, welche die Produktion von ApoC3 oder Angiopoietin-like Protein 3 (ANGPTL3) hemmen und dadurch die Triglyzeride senken.
Bei der akuten Pankreatitis konnte die Kausalität in genetischen Studien gezeigt werden, besonders am Beispiel der familiären Chylomikronämie.10 Daten aus Interventionsstudien oder Tiermodellen dazu existieren aber nicht. «Meiner Meinung nach ist die Hypertriglyzeridämie ein unterschätzter Risikofaktor für die Entstehung einer akuten Pankreatitis – neben Alkohol und Gallenwegserkrankungen wahrscheinlich einer der wichtigsten», so der Experte.
Eine ähnliche Evidenzlage zeigt sich beim Diabetes mellitus: Genetische Studien legen Kausalität nahe, Daten aus Interventionsstudien oder Tiermodellen gibt es nicht. In Bezug auf die Fettleber gibt es ebenfalls keine Interventionsstudien, in genetischen Studien und Tiermodellen zeigt sich die Beziehung genabhängig.
Lipoprotein(a) – starke genetische Hinweise, fehlende RCT-Daten
Auch hohe Spiegel von Lipoprotein(a) (Lp[a]) erhöhen das Risiko für atherosklerotische Erkrankungen. Sowohl genetische Studien bei Menschen als auch genetische Tiermodelle sprechen für Kausalität. Studien zur Wirksamkeit der Lp(a)-Senkung in der Atheroskleroseprävention laufen. Auch die Risiken für Aortenstenose, Herzinsuffizienz und gleichzeitige Mortalität sind bei hohem Lp(a) erhöht. Diese Assoziationen sind aus genetischer Sicht kausal.
Das Risiko für venöse Thrombosen, für das bei Kindern Lp(a) als Risikofaktor gilt, liess sich bei Erwachsenen nicht mit den Lp(a)-Werten in Zusammenhang bringen – weder in epidemiologischen noch in genetischen Studien. Auch hier liegen keine Daten aus Interventionsstudien oder Tiermodellen vor.
Bezüglich des Diabetesrisikos konnte in epidemiologischen Studien eine inverse Beziehung zu Lp(a) beobachtet werden – hohe Werte schützten also. Das war auch genetisch nachweisbar: Veränderungen im LPA-Gen, die das Risiko für eine Atherosklerose erhöhten, waren im Hinblick auf einen Diabetes mellitus protektiv.
«In genomweiten Assoziationsstudien, die die wichtigsten Gene heraussuchen, die für einen Herzinfarkt prädisponieren, liegt das LPA-Gen auf Platz 2, bei Aortenstenose sogar auf Platz 1», ergänzte von Eckardstein.
In Mendel’schen Randomisierungsstudien konnten auch für den ischämischen Hirninfarkt Hinweise auf einen Kausalzusammenhang generiert werden, nicht aber für den hämorrhagischen Hirninfarkt und auch nicht für Demenzerkrankungen.11
«Lp(a) ist ein LDL-ähnliches Molekül, das ganz ähnliche Mechanismen mit diesem teilt und somit proatherogenetisch wirkt. Zusätzlich verfügt es durch das Apo(a) vermutlich über die Fibrinolyse hemmende Eigenschaften und wirkt so wohl zusätzlich prothrombotisch. Zudem finden sich die oxidierten Phospholipide zumeist im Lp(a), was die proinflammatorischen Effekte auslöst»,12 erklärte der Referent den möglichen Pathomechanismus. Therapeutisch seien injizierbare RNA-basierte Therapien (Antisense-Oligonukleotide und small interfering RNA, siRNA) derzeit in der klinischen Erprobung, die das Lp(a) um 80–90% senken könnten, ebenso orale kleine Moleküle, die ebenfalls Lp(a) senken.13
HDL-Cholesterin – wirklich immer «das Gute»?
«Das HDL-C hat eine inverse Beziehung zum Herzinfarkt, aber nur bis in einen mittleren Bereich. Hohe HDL-C-Konzentrationen bedeuten keine Risikominderung. Sie erhöhen sogar das Mortalitätsrisiko», erklärte von Eckardstein. «In klinischen Studien hat eine HDL-C-Erhöhung keine kardiovaskulären Ereignisse verhütet.» Das könne möglicherweise an den verwendeten Medikamenten liegen: Durch die sehr stark HDL-erhöhende Wirkung der hier eingesetzten CETP-Inhibitoren könnte es zu Werten im oberen Risikobereich der parabolischen HDL-Kurve gekommen sein, besonders bei denjenigen, die zu Studienbeginn bereits im mittleren, risikoarmen HDL-Wertebereich lagen. Genetische Studien konnten zudem keine Hinweise auf einen kausalen Zusammenhang liefern. In Tiermodellen waren die Zusammenhänge genabhängig.
«Was wir jedenfalls epidemiologisch ganz klar beobachten konnten, ist, dass ab einer bestimmten Höhe des HDL-C das Herzinfarktrisiko nicht weiter sinkt, die Mortalität dagegen aber ansteigt. Das HDL medikamentös möglichst weit nach oben zu bringen, so wie es durch die CETP-Inhibitoren in Studien gemacht wurde, kann daher nicht das Ziel sein. Und auch Fibrate und Nikotinsäure sind keine geeigneten Medikamente, da sie eher wenig Effekt auf das HDL-C haben. Sie wirken stärker auf die Triglyzeride.»
Auch zu verschiedenen anderen Krankheiten besteht ein beobachtbarer Zusammenhang zu HDL. So haben Menschen mit niedrigen HDL-Werten ein erhöhtes Diabetesrisiko. Der Zusammenhang zeigte sich in Interventions- und genetischen Studien sowie im Tiermodell kausal. Daher hat HDL auch in fast alle Risikoscores zum Diabetes als Parameter Eingang gefunden.
Die Beziehung von HDL-C und chronischen Nierenerkrankungen sowie Infektionen ist parabolisch. Daten aus genetischen Studien und Tiermodellen sprechen für Kausalität. Interventionsstudien existieren hier bisher nicht. Auch ein inverser Zusammenhang zwischen HDL-C und einigen Autoimmunerkrankungen, z.B. chronisch-entzündlichen Darmerkrankungen, wurde beobachtet und Daten aus Tiermodellen legen eine mögliche Kausalität nahe. Jedoch gibt es hier weder Interventions- noch genetische Studien.
Dass hohe HDL-Werte nicht immer protektiv wirken, zeigt auch das bei Menschen mit hohem HDL-C erhöhte Risiko für eine altersbedingte Makuladegeneration. Auch liefern genetische Studien und Tiermodelle Hinweise auf Kausalität. Hinsichtlich des Alzheimerrisikos wurde wiederum ein parabolischer Zusammenhang beobachtet, für den genetische Studien heterogene Daten lieferten. Ergebnisse aus Tiermodellen lassen aber einen Kausalzusammenhang zwischen niedrigem HDL-C und Demenzrisiko plausibel erscheinen.14
Für das Scheitern von HDL-C-Interventionen in der kardiovaskulären Prävention und Therapie gab von Eckardstein abschliessend mehrere mögliche Begründungen an: Neben einer eventuell tatsächlich fehlenden Kausalität komme auch eine reverse Kausalität infrage. Das bedeutet, dass die niedrigen HDL-C-Spiegel Folge bestimmter Erkrankungen sein könnten und nicht umgekehrt. Denkbar wäre auch, dass HDL-C nur ein pathophysiologisch unbeteiligter «Zuschauer» ist, der von anderen Faktoren, die ein erhöhtes Atheroskleroserisiko bedingen, quasi im Nebenschauplatz erniedrigt wird. «Vermutlich messen wir mit HDL-C auch einfach den falschen Biomarker. Denn es gibt zahlreiche Subklassen von HDL, die wiederum hunderte von Proteinen und Lipiden enthalten. Dabei variiert aber die Anzahl der Moleküle pro Partikel stark: Enthält ein HDL-Partikel etwa 1000 Moleküle Cholesterin, das an der biologischen Funktion des HDL gar nicht beteiligt ist, kommen die funktionsbestimmenden Proteine und Lipide um ein Vielfaches seltener vor. Das Sphingosin-1-Phosphat zum Beispiel ist nur in etwa jedem zehnten HDL-Partikel mit einem Molekül enthalten. Das Messinstrument ‹Cholesterin›, das wir momentan einsetzen, ist daher völlig ungeeignet, die biologische Wirksamkeit von HDL zu messen», schloss er seine Überlegungen.
Quelle:
1. AGLA-Kurs Klinische Lipidologie, 18.–19. Januar 2024, Zürich
Literatur:
1 Benn M et al.: From genome-wide association studies to Mendelian randomization: novel opportunities for understanding cardiovascular disease causality, pathogenesis, prevention, and treatment. Cardiovasc Res 2018; 114: 1192-208 2 McPherson et al.: Genetics of coronary artery disease. Circ Res 2016; 118: 564-78 3 Ference BA et al.: Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017; 38: 2459-72 4 Borén J et al.: Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2020; 41: 2313-30 5Tokgözoğlu L et al.: The dawn of a new era of targeted lipid-lowering therapies. Eur Heart J 2022; 43: 3198-208 6 Preiss D et al.: Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA 2011; 305: 2556-64 7 Perego C et al.: Cholesterol metabolism, pancreatic β-cell function and diabetes. Biochim Biophys Acta Mol Basis Dis 2019; 1865: 2149-56 8 Tragante V et al.: Harnessing publicly available genetic data to prioritize lipid modifying therapeutic targets for prevention of coronary heart disease based on dysglycemic risk. Hum Genet 2016; 135: 453-67 9 Sun L et al.: Causal associations of blood lipids with risk of ischemic stroke and intracerebral hemorrhage in Chinese adults. Nat Med 2019; 25: 569-74 10 Pedersen SB et al.: Nonfasting mild-to-moderate hypertriglyceridemia and risk of acute pancreatitis. JAMA Intern Med 2016; 176: 1834-42 11 Larsson SC et al.: Lipoprotein(a) in alzheimer, atherosclerotic, cerebrovascular, thrombotic, and valvular disease: Mendelian Randomization Investigation. Circulation 2020; 141: 1826-8 12 Tsimikas S: A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol 2017; 69: 692-711 13 Nicholls SJ et al.: Muvalaplin, an oral small molecule inhibitor of lipoprotein(a) formation: a randomized clinical trial. JAMA 2023; 330: 1042-53 14 von Eckardstein A et al.: HDL – quo vadis. Dtsch Med Wochenschr 2023; 148: 627-35
Das könnte Sie auch interessieren:

Mechanische Kreislaufunterstützung im Infarkt-bedingten kardiogenen Schock
Der Infarkt-bedingte kardiogene Schock (AMI-CS) ist trotz der enormen Fortschritte in der interventionellen Versorgung des akuten Myokardinfarktes in den vergangenen Jahrzehnten ...
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...