
Neue Therapien bei Muskelerkrankungen
Die Diagnose einer spinalen Muskelatrophie (SMA) bei den großteils sehr jungen Patienten stellt deren Eltern vor eine gewaltige Herausforderung, ist sie doch verbunden mit einem zunehmenden Verlust der Mobilität bis hin zu einem Leben im Rollstuhl und einer verkürzten Lebenserwartung. Neue Therapien mit beeindruckenden Ergebnissen schüren nun hohe Erwartungen bei Betroffenen und Medizinern.
Muskelerkrankungen zählen zu den seltenen Erkrankungen. Von den circa 20000 Betroffenen in Österreich sind mehr als die Hälfte Kinder und Jugendliche. Prim. Univ.-Prof. Dr. Günther Bernert, Neuropädiater und Vorstand der Abteilung für Kinder- und Jugendheilkunde am Kaiser-Franz-Josef-Spital mit Gottfried von Preyer’schem Kinderspital am SMZ Süd, und Prof. Wolfgang Schmidt, Neuromuskuläre Forschungsabteilung an der Medizinischen Universität Wien, berichteten bei den Pädiatrietagen in Venedig über die neuesten Entwicklungen bei den Therapieoptionen für spinale Muskelatrophie.
Spinale Muskelatrophie
SMA ist die zweithäufigste autosomal rezessive Muskelerkrankung im Kindesalter mit einer Inzidenz von ca. 1:7000 bis 10000 Lebendgeborenen. Sie ist zudem die häufigste Todesursache im Säuglings- und Kleinkindalter.1Leitsymptom der SMA ist das sogenannte „Floppy-Infant-Syndrom“, die infantile Muskelhypotonie. Bernert beschrieb den typischen SMA-Patienten in der pädiatrischen Klinik als Säugling jünger als sechs Monate, der seine Beine in Rückenlage nicht aufgestellt halten kann, mit auffälligem Atemtypus und eingeschränkter Kopfkontrolle, aber aufmerksam und kognitiv nicht beeinträchtigt.
Die Ursache für diese Erkrankung liegt in einem defekten Protein SMN („survival of motor neuron“), das für die Funktion der Motorneuronen essenziell ist. Bei einem Defekt im SMA-Protein kommt es zum zunehmenden Abbau von Alpha-Motoneuronen im Rückenmark. Progressive Muskelatrophie und der Verlust von Mobilität und Motorik sind die Folge. Das SMN-Protein wird von zwei Genen codiert: Das SMN1-Gen liefert 90–95% des Proteins, das homologe Gen SMN2 fungiert unterstützend, liefert aber nur 5–10% der Proteinmenge. Grund dafür ist der Austausch eines Nukleotids in Exon 7, wodurch es für den weiteren Ablauf verloren geht. SMN2 wird nicht korrekt prozessiert, was die reduzierte Produktion von funktionellem SMN-Protein zur Folge hat. SMN2 kann somit einen Defekt in SMN1 nicht vollwertig ersetzen.
Der homozygote Verlust von SMN1 resultiert in der Manifestation von SMA. SMN2, das im Genom des Patienten in unterschiedlicher Kopienzahl vorliegt, beeinflusst die Ausprägung der klinischen Manifestation. Man unterscheidet dabei vier verschiedene SMA-Typen (Tab. 1). SMA Typ 1 ist dabei mit ca. 60% der häufigste Typus und hat den schwersten Verlauf. „Wir wissen, dass unbehandelt alle Kinder mit SMA Typ 1 bis zum Alter von 24 Monaten verstorben sein werden“, sagte Bernert.Aber auch wenn bei der Diskussion über die Behandlung der SMA der Fokus auf diesen schwer erkrankten Patienten liege, dürfe man nicht außer acht lassen, dass auch bei Patienten mit einer leichteren Form der Erkrankung (Typ 3 und 4) ein progredienter Verlauf vorprogrammiert sei. Eine Stabilisierung sei bei keinem SMA-Typus zu erwarten, gab Bernert zu bedenken. Daher muss jeder Patient individuell betrachtet werden. Für einen Patienten, dessen Gehfunktion stark eingeschränkt ist, der aber im Rollstuhl gut integriert ist, sei die Erhaltung der Funktion der oberen Extremitäten wesentlich wichtiger als nur die Funktion der Beine.
„Standards of care“ sind unerlässlich
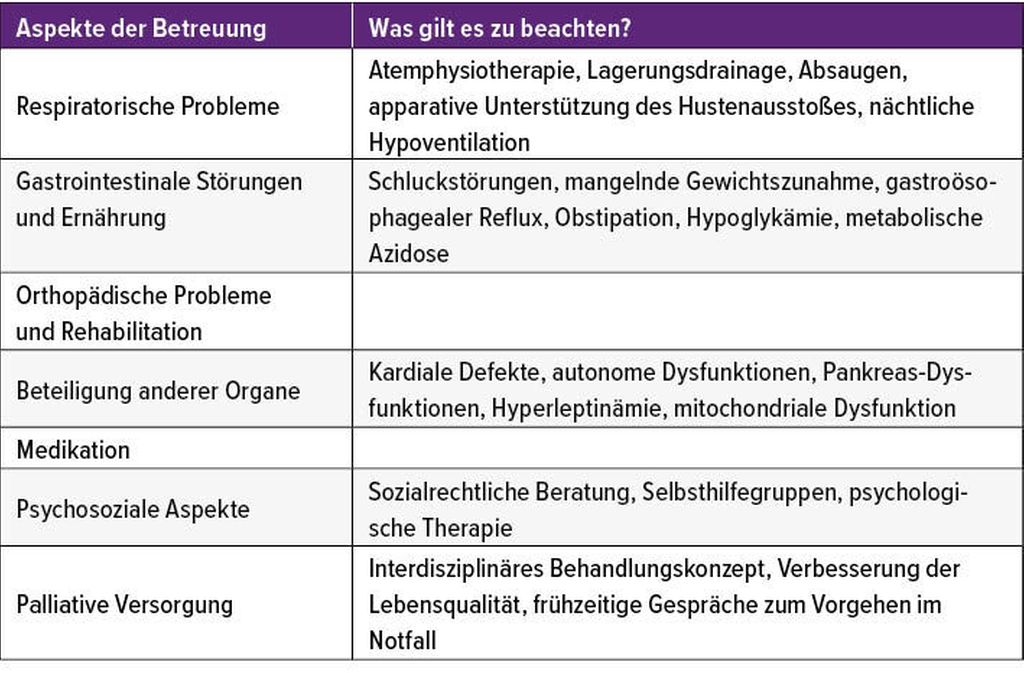
Tab. 2: „Standards of care“ für SMA-Patienten sind vielschichtig
Die „standards of care“ sind in der Betreuung von SMA-Patienten eine wichtige Säule. Aufgrund der Komplexität der Erkrankung erfordert die umfassende, fächerübergreifende medizinische Betreuung von SMA-Patienten ein Team aus verschiedenen Bereichen (Tab. 2).2 Abgestimmt auf den Verlauf des Gesundheitszustandes des Patienten müssen diese Standards immer wieder evaluiert und angepasst werden. „Wichtig ist die proaktive Antizipation der nächsten Schritte. ‚Standards of care‘ und medikamentöse Therapie müssen immer zusammengebracht werden. Denn die beste dieser Therapien kann nicht ihr volles Potenzial entfalten, wenn nicht auch gleichzeitig die ‚standards of care‘ angewendet werden. Deshalb sind auch die neuen Verlaufsstudien unter den neuen Therapien immer darauf abgestimmt, dass die ‚standards of care‘ angewendet werden“, berichtete Bernert.
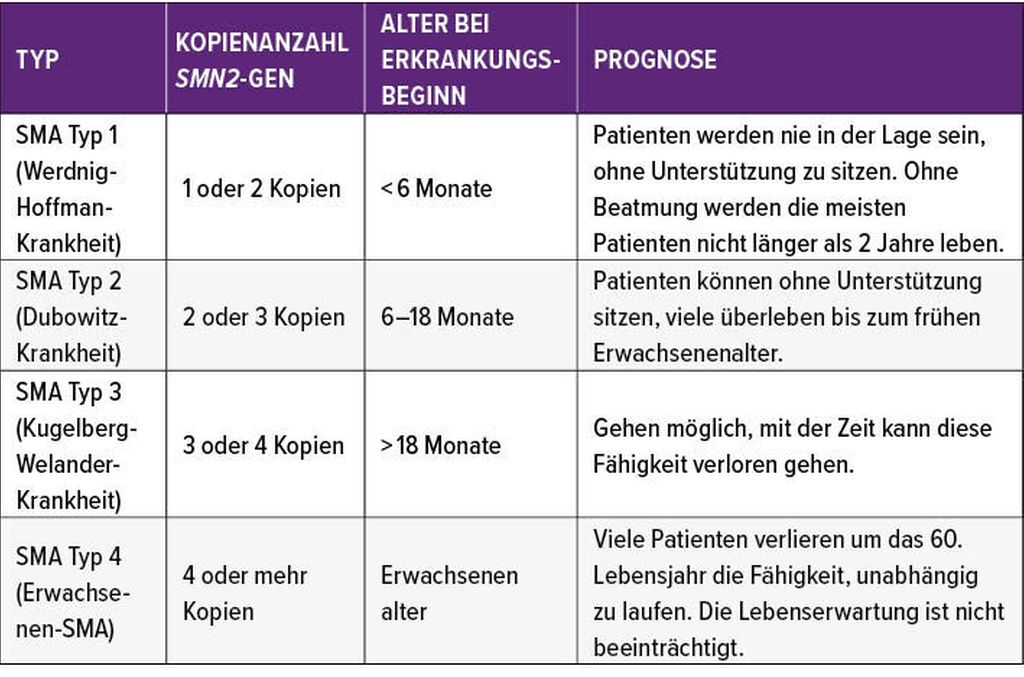
Tab. 1: Ausprägung der SMA-Erkrankung abhängig von der Kopienzahl von SMA-2
Neue Ansätze zur Therapie der SMA
Nusinersen (SpinrazaTM) wurde 2016 als erste ursächliche Therapie für SMA von der EMA zugelassen. Sie zielt darauf ab, SMN2 „fit“ zu machen. Es soll mehr funktionelles SMN-Protein produzieren und so das mutierte SMN1-Gen nahezu vollwertig ersetzen. Dabei wird ein komplementärer RNA-Strang (Antisense-RNA) in die Zellen eingebracht, der sicherstellt, dass Exon 7 nicht ausgelassen wird. „Das für diesen Prozess notwendige Molekül ist kurz, stabil, passiert aber nicht die Blut-Hirn-Schranke. Nusinersen hat eine Halbwertszeit von circa sechs Monaten. Dies bedingt die regelmäßige Verabreichung in Form einer intrathekalen Injektion“, sagte Schmidt. Im ersten Jahr der Therapie sind im Sinn einer Aufsättigung sechs Injektionen vorgesehen, danach drei pro Jahr.
„Dieses Medikament hat schon in den ersten Studien große Erfolge gezeigt, die in den nachfolgenden Real-Life-Studien bestätigt wurden“, berichtete Bernert. „Die Geschwindigkeit und das Ausmaß des Ansprechens auf die Therapie ist abhängig vom Alter der Patienten bei Therapiebeginn, je früher desto besser. Besonders profitierten jene Kinder, die präsymptomatisch behandelt wurden, also Geschwisterkinder, wo pränatale Diagnostik gemacht wurde, und Kinder aus neonatalen Screeningprogrammen.“ Am Preyer’schen Kinderspital werden rund 20 Patienten im Alter zwischen zwei Monaten und 15 Jahren mit Nusinersen behandelt. Bei allen Patienten konnte zumindest eine Stabilisierung erreicht werden, bei vielen Patienten sogar eine Funktionsverbesserung.
Hoffnung Gentherapie
Eine andere Strategie verfolgt eine Therapie, die mittlerweile ebenfalls kurz vor der Zulassung steht. Bei der Genersatztherapie mit AVXS-101 (Onasemnogene Abeparvovec, ZolgensmaTM) wird ein rekombinantes, fehlerfreies SMN1-Gen mithilfe eines „adeno-associated virus“ (Serotyp AAV9, mit einem besonderen Tropismus für das zentrale Nervensystem) in den Körper eingebracht. „Es vermehrt sich dort ohne Modifikation oder Integration in die DNA des Patienten und liegt episomal als zirkuläres, eigenständiges Molekül in der Zelle des Patienten vor. So stellt es eine dauerhafte und zielgerichtete Quelle der SMN-Proteinexpression sicher“, erklärte Schmidt. „Diese Partikel sind so klein, dass sie die Blut-Hirn-Schranke passieren können. Das bedeutet, dass im Gegensatz zu einer Antisense-RNA eine einmalige Gabe i.v. möglich ist. Eine zweite Gabe ist wegen der zu erwartenden hohen Titer gegen das Virus nicht möglich. Aus dem gleichen Grund ist diese Form der Therapie bei Erwachsenen so nicht möglich.“
In die Zulassungstherapie für Onasemnogene Abeparvovec wurden 12 Patienten mit SMA Typ 1 eingeschlossen (Stand 31. Mai 2019). Alle Studienteilnehmer hatten zwei SMA2-Kopien und waren bei Therapiebeginn symptomatisch. „Beim 2-Jahres-Follow-up waren alle zwölf Patienten am Leben und ohne permanente Beatmung. Elf von ihnen konnten eigenständig sitzen, zwei Kinder krabbelten, konnten sich an Gegenständen hochziehen, stehen und selbstständig gehen. Wir sehen also auch hier eine vielversprechende Therapie, deren Erfolg wir antizipieren können“, resümierte Bernert.
Die Nebenwirkungen dieser Gentherapie waren sehr gut zu managen. Transiente Transaminasenerhöhungen wurden durch die Begleittherapie von Steroiden vor und nach der Injektion sehr gut managebar. Andere Komplikationen waren vorhanden und in Ausmaß und Schweregrad von der Grunderkrankung abhängig. Sie waren aber nicht direkt von der Therapie abhängig.
Bericht: Dr. Gabriele Senti
Quelle:
Pädiatrietage der Österreichischen Gesellschaft für Kinder- und Jugendheilkunde, 22. November 2019, Venedig
Literatur:
1 Verhaart IEC et al.: Orphanet J of Rare Diseases 2017; 12: 124 2 Borell S et al.: Monatsschr Kinderheilk 2015; 163: 1293-304
Das könnte Sie auch interessieren:

Menschen mit Demenz: Was beeinflusst deren Überleben nach Diagnosestellung?
Verschiedenste Faktoren beeinflussen die Überlebenszeit nach einer Demenzdiagnose. Das Wissen um Risikofaktoren zum Zeitpunkt der Diagnose einer Demenzerkrankung oder in deren Verlauf ...

Alzheimer: Was gibt es Neues in der Biomarker-Entwicklung?
Schätzungen zufolge leben in Österreich 115000 bis 130000 Menschen mit einer Form der Demenz. Eine Zahl, die sich bis zum Jahr 2050 verdoppeln wird.1 Antikörper-Wirkstoffe könnten in der ...

Kappa-FLC zur Prognoseabschätzung
Der Kappa-freie-Leichtketten-Index korreliert nicht nur mit der kurzfristigen Krankheitsaktivität bei Multipler Sklerose, sodass er auch als Marker zur Langzeitprognose der ...