
L’amyloïdose comme cause d’insuffisance cardiaque à FE préservée
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Au cours des dernières années trois changements de paradigmes ont eu lieu dans le domaine de l’amyloïdose cardiaque: 1) une approche majoritairement basée sur l’imagerie et sans biopsie permet de diagnostiquer le type d’amyloïdose le plus prévalent (amyloïdose à transthyrétine); 2) il a été démontré qu’une proportion considérable de patients avec une insuffisance cardiaque à FE préservée souffrent d’une amyloïdose cardiaque; 3) un traitement modifiant le cours de la maladie a été approuvé. Une bonne collaboration entre médecin généraliste, cardiologue et dans certains cas hématologue permet d’identifier, caractériser et prendre en charge les patients atteints de cette maladie mortelle.
Keypoints
-
L’amyloïdose cardiaque (AC) de type ATTRwt n’est pas une entité rare et doit être suspectée en cas d’insuffisance cardiaque à fraction d’éjection préservée.
-
La scintigraphie osseuse/SPECT au 99mTc-DPD permet de diagnostiquer l’AC ATTR après exclusion formelle d’une gammapathie monoclonale.
-
De nouveaux traitements de l’AC ATTR ralentissent la progression de la maladie.
La prévalence de l’insuffisance cardiaque augmente continuellement. Il est estimé que le nombre de cas en Suisse s’élève actuellement à 150000, dont près de la moitié souffre d’une insuffisance cardiaque à fraction d’éjection préservée (HFpEF). Cette dernière est définie par une fraction d’éjection du ventricule gauche (FEVG) de >50% en présence de symptômes ou de signes d’insuffisance cardiaque et de la mise en évidence d’anomalies cardiaques compatibles avec une dysfonction diastolique/augmentation de la pression de remplissage du VG.1 L’élévation de la pression de remplissage est démontrée soit par les caractéristiques échocardiographiques, soit par mesures invasives ou encore par l’augmentation des peptides natriurétiques (BNP/nt-proBNP) au niveau sanguin. L’HFpEF regroupe une population hétérogène, souvent âgée, avec des comorbidités multiples, ce qui rend l’identification de l’étiologie précise difficile. Une maladie coronarienne, l’hypertension artérielle (HTA), un diabète ou une obésité peuvent en être la cause, et le sont fréquemment de façon combinée. Le pronostic de ce type d’insuffisance cardiaque est mauvais et les traitements médicamenteux à disposition sont souvent décevants. Il est d’autant plus important de chercher chez chaque patient une étiologie précise de la HFpEF afin de pouvoir anticiper des complications et, si indiqué, de débuter une des rares thérapies ciblées.
L’amyloïdose cardiaque (AC) est une étiologie de la HFpEF qui se distingue par son caractère évolutif menant dans tous les cas vers des complications telles que troubles de la conduction, arythmies, insuffisance cardiaque terminale. Le diagnostic précoce et précis permet de diriger le patient vers une prise en charge spécialisée et potentiellement vers un traitement spécifique capable de ralentir la progression de la maladie.
Qu’est-ce que l’amyloïdose cardiaque?
L’AC s’inscrit dans le contexte d’une maladie systémique infiltrative dont les dépôts myocardiques, composés de protéines, mènent à une cardiomyopathie restrictive. Les protéines sont soit des chaînes légères d’immunoglobulines (produites par un clone plasmocytaire dans le cadre d’une gammapathie monoclonale, la maladie est alors appelée amyloïdose light-chain ou AL), soit de la transthyrétine (TTR), une protéine de transport plasmatique synthétisée par le foie (amyloïdose ATTR). La transthyrétine est mutée et défectueuse dans le cas de l’amyloïdose ATTRm (TTR muté), ou non mutée dans le cas de l’amyloïdose ATTRwt (TTR wild type). Les autres formes d’amyloïdose, notamment l’amyloïdose AA (dépôt d’amyloïde A) qui est associée aux maladies inflammatoires chroniques, ne causent que très rarement une cardiomyopathie. Les fibrilles d’amyloïde se déposent dans de nombreux organes sans nécessairement de traduction clinique. Dans l’AL c’est en général l’atteinte rénale et/ou cardiaque qui est au premier plan, tandis que pour l’ATTR, ce sont les dégâts au niveau ligamentaire/tendineux du système nerveux périphérique et/ou du cœur. La forme ATTRwt en est de loin la plus fréquente. Elle survient en général chez le sujet de >65 ans et plus fréquemment chez l’homme, tandis que les autres types d’AC peuvent survenir à un jeune âge et à une fréquence comparable dans les deux sexes.3
Les techniques d’imagerie actuelles permettent un diagnostic fiable de la maladie, et leur utilisation systématique a aidé à démontrer que la forme ATTRwt est nettement plus prévalente que ce qui était initialement suspecté. Des études montrent que dans la population avec une HFpEF la prévalence de l’amyloïdose cardiaque est de plus de 10%.2−4 Un constat similaire est fait chez des patients âgés qui se présentent avec une sténose aortique.5,6
Chez qui rechercher uneamyloïdose cardiaque?
Certaines caractéristiques cliniques et paracliniques ont été identifiées comme «red flags», permettant au médecin généraliste ou au cardiologue de suspecter une amyloïdose cardiaque, notamment chez des patients de >65 ans avec une HFpEF, sténose aortique ou hypertrophie ventriculaire gauche:
-
syndrome du tunnel carpien bilatéral
-
canal lombaire étroit
-
polyneuropathie périphérique ou dysautonomie idiopathique
-
tension artérielle (TA) normale ou hypotension orthostatique si anciennement hypertendu
-
ECG avec microvoltages, absence d’ondes R en précordial ou pseudo-ondes Q (sans notion d’infarctus) (Fig.1)
-
Echocardiographie avec hypertrophie ventriculaire gauche (septum >12mm), dysfonction diastolique, dilatation atriale, discret épanchement péricardique (Fig. 2) et diminution de la contraction longitudinale avec épargne apicale (Fig. 3)

Fig. 1: ECG en fibrillation atriale d’un patient de 79 ans avec amyloïdose ATTRwt. Noter la présence d’un hémibloc antérieur gauche, des ondes Q en II, II, avF (pseudo-infarctus), et une absence de progression de l’onde R de V1 – V6)

Fig. 2: Echocardiographie transthoracique. Vue apicale 4 cavités: hypertrophie concentrique bi-ventriculaire (flèches bleues) et dilatation biatriale (*)
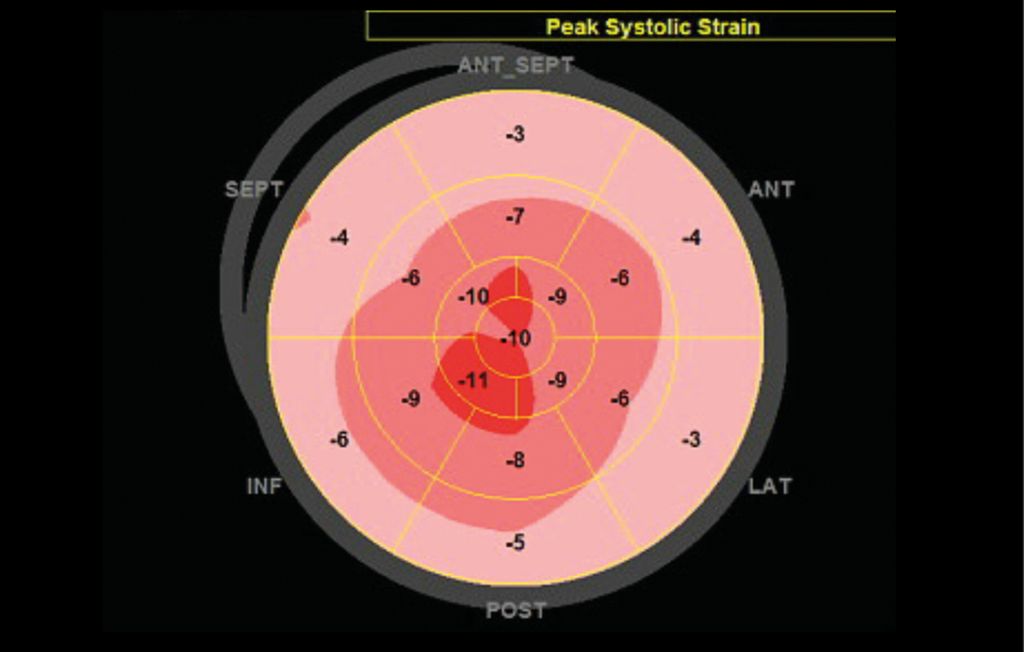
Fig. 3: Diagramme en cible («bull’s eye»). Ce diagramme montre les valeurs de strain longitudinal (déformation longitudinale) systolique du ventricule gauche pour chaque segment myocardique. La valeur indique le pourcentage de raccourcissement des fibres longitudinales en systole. On note une réduction sévère du strain dans les segments basaux et moyens (périphérie du diagramme, couleur rose pâle) alors que le strain des segments apicaux est moins diminué (centre du diagramme, couleur rouge); cette distribution est caractéristique de l’amyloïdose cardiaque
L’algorithme diagnostique
Le «Swiss Amyloidosis Network (SAN)», un groupe de travail multidisciplinaire national, a publié des recommandations très détaillées en 20217, avec notamment une approche diagnostique standardisée qui, dans les grandes lignes, adopte l’algorithme établi au niveau international (Fig.4).
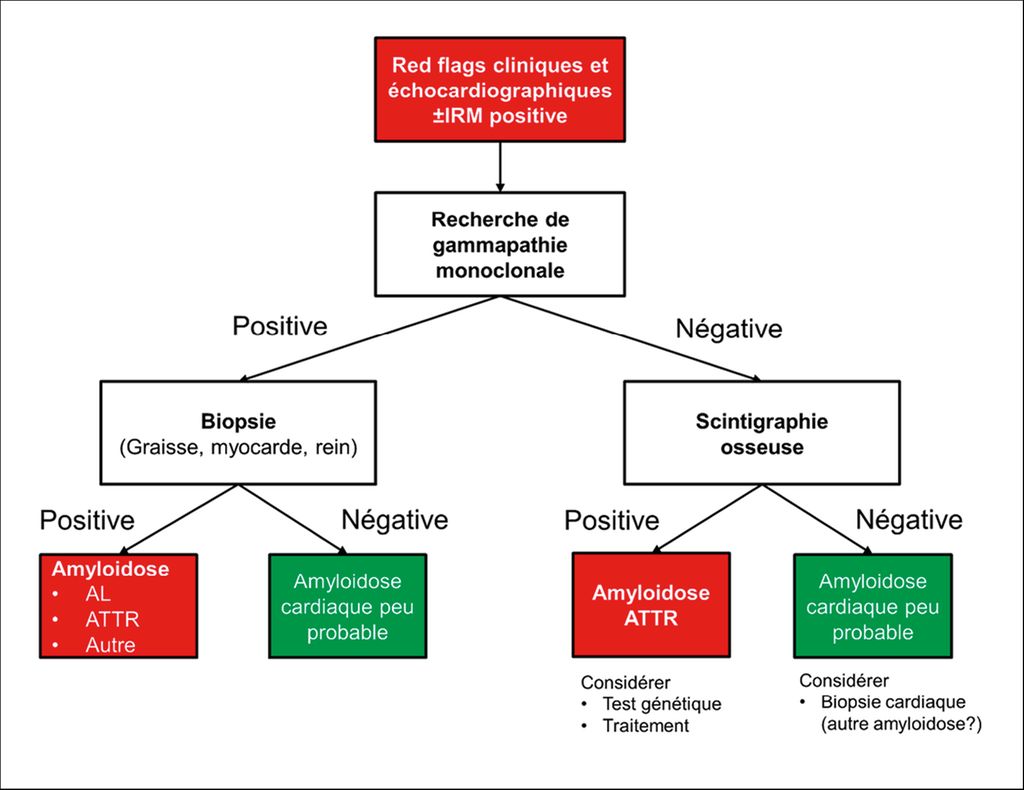
Fig 4: Algorithme diagnostic de l’amyloïdose cardiaque (adapté d’après Hugelshofer et al.)23

Fig 5: IRM cardiaque. Vue en court axe en mi-ventriculaire en séquence de rehaussement tardif. Dans cette séquence, le myocarde normal doit apparaître uniformément noir. On note chez ce patient un rehaussement circonférentiel à prédominance sous-endocardique (flèches bleues), touchant également le ventricule droit (flèches jaunes)
Contrairement à l’ETT et la scintigraphie/SPECT, l’IRM cardiaque n’est pas indispensable pour le diagnostic mais complémentaire à l’échocardiographie, surtout en cas de doute avant de suivre tout l’algorithme diagnostique. Elle permet de préciser la morphologie et la fonction ventriculaire systolique, mais surtout de démontrer l’infiltration du myocarde par les fibrilles amyloïdes, qui se manifeste par une prise de contraste sur les séquences de rehaussement tardif (RT). L’étendue du RT reflète la sévérité de l’infiltration (Fig. 5).8 Cette dernière, cependant, peut être quantifiée de manière plus précise par la mesure du volume extracellulaire myocardique, en utilisant une séquence de cartographie T1. Un volume extracellulaire >40% indique ainsi une infiltration sévère, hautement suggestive d’un diagnostic d’amyloïdose.9
L’étape diagnostique clé est d’exclure une origine néoplasique (type AL) par la recherche active d’une gammapathie monoclonale dans le sang et les urines (immunofixation des protéines et dosage des chaînes légères libres). En cas d’évidence pour une gammapathie monoclonale, il est cependant indispensable de confirmer la présence de dépôts amyloïdes par la biopsie d’un organe atteint. En effet, dans une population âgée, la prévalence de la gammapathie monoclonale de signification indéterminée (MGUS) étant élevée (5,3% à >70 ans),10 la combinaison d’une amyloïdose ATTRwt et d’une MGUS pourrait facilement être confondue avec une amyloïdose AL en l’absence de preuve histologique. Ceci risquerait alors de conduire à l’administration de traitements inappropriés.
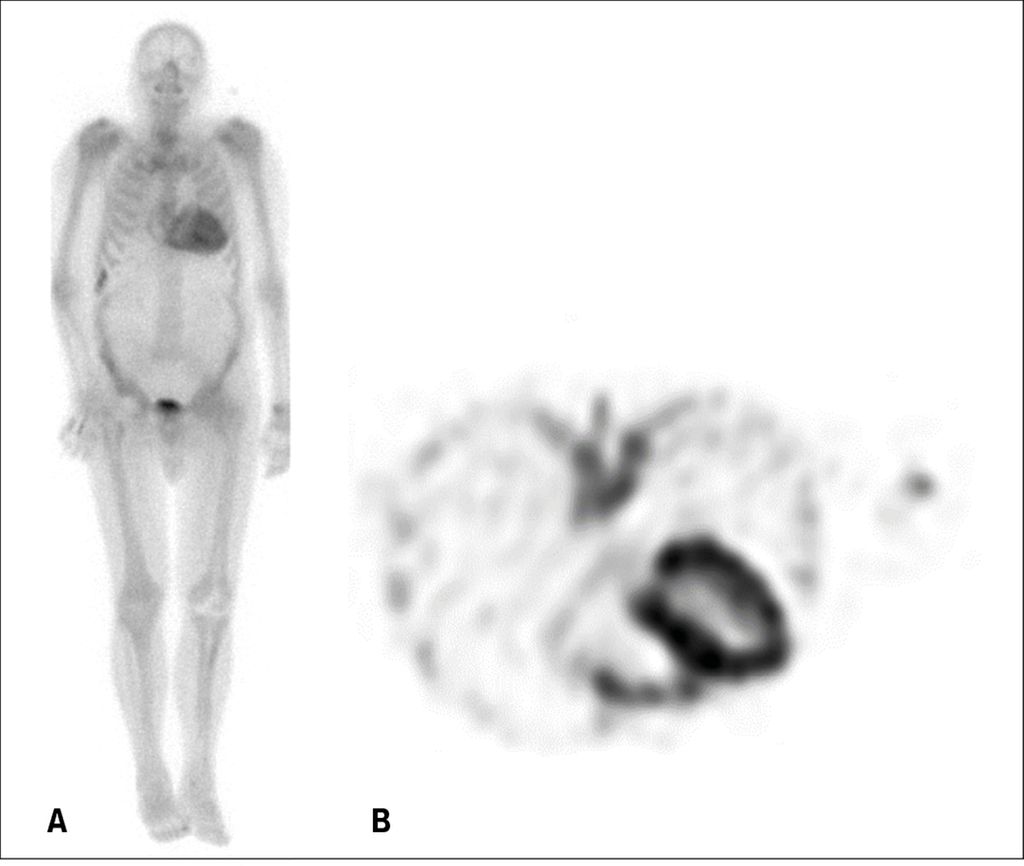
Fig 6: Scintigraphie osseuse/SPECT au 99mTc-DPD (dipropionate de béclométhasone) chez un patient avec amyloïdose cardiaque ATTR. A) Image planaire; B) Image de SPECT
La scintigraphie du corps entier avec SPECT (Single Photon Emission Computer Tomography) utilisant un marqueur osseux (en Suisse souvent 99mTc-DPD), fait partie intégrante du diagnostic de l’amyloïdose ATTR. En effet, dans l’amyloïdose, le traceur osseux se lie aux micro-calcifications contenues dans les fibrilles d’amyloïde et s’accumule dans le myocarde (Fig. 6). Cette technique est donc très sensible pour détecter l’AC de type ATTR,11 même si sa spécificité n’est pas parfaite, car la scintigraphie est faiblement positive dans environ un tiers des cas d’amyloïdose AL.12 L’examen peut être faussement positif en cas d’infarctus étendu récent, si une scintigraphie simple (images planaires sans SPECT) est effectuée et l’accumulation du traceur dans le «blood pool» est confondu avec un «uptake» au niveau myocardique, ou dans les rares cas de cardiomyopathie toxique à la chloroquine. Toutefois, après l’exclusion soigneuse d’une gammapathie monoclonale, une scintigraphie osseuse positive au niveau myocardique est considérée comme un diagnostic d’une amyloïdose ATTR. À l’inverse, en cas de négativité de la scintigraphie et en l’absence de gammapathie monoclonale, une AC sera virtuellement exclue. Dans ces cas, la biopsie myocardique n’est réservée qu’aux cas dont la suspicion clinique d’amyloïdose reste élevée.
Même s’il est inhabituel qu’une amyloïdose familiale (ATTRm) se révèle chez un patient de >65 ans, le testing génétique est conseillé chez tous les patients avec une AC ATTR au moment du diagnostic, indépendamment de leur âge. Ceci se justifie par le fait que la présence d’une mutation comporte des implications directes pour les membres de la famille au premier degré.
Pronostic
De façon générale, le pronostic de toute amyloïdose systémique est fortement influencé par la présence ou non d’une infiltration cardiaque. Quand celle-ci est présente, le pronostic est alors dépendant du type d’amyloïdose. La survie médiane sans traitement est de 5,4 mois pour une AC de type AL versus 60 mois pour une AC de type TTR.8,13 Bien que l’estimation individuelle du pronostic se base essentiellement sur le dosage des biomarqueurs cardiaques (troponines T et nt-proBNP), la sévérité de la dysfonction longitudinale du ventricule gauche en échocardiographie14 et l’étendue du RT en IRM cardiaque8 ont montré une valeur pronostique indépendante.
Traitement
Les traitements peuvent être divisés en deux groupes: le traitement symptomatique de l’insuffisance cardiaque d’une part, et les traitements ayant pour but de ralentir la progression de la maladie et d’améliorer le pronostic d’autre part («disease modifying treatments»).
Traitements symptomatiques
L’AC est un modèle de cardiopathie restrictive caractérisée par une petite cavité ventriculaire gauche peu compliante. De ce fait une contraction atriale conservée joue un rôle important dans l’optimisation du remplissage ventriculaire malgré la dysfonction diastolique. Pour soulager les symptômes, dont la dyspnée est au premier plan, on doit donc s’appliquer avant tout à optimiser la volémie, et à maintenir un rythme sinusal. Le myocarde rigide n’ayant plus la capacité de s’adapter à une variation de la précharge, un traitement diurétique ajusté de façon régulière permettra de garder un remplissage ventriculaire adéquat. Dans les cas avancés, la marge thérapeutique est toutefois étroite, avec des oscillations fréquentes entre surcharge volémique et insuffisance rénale, rendant l’équilibre difficile. L’infiltration des oreillettes et la surcharge en pression mènent systématiquement à une dilatation atriale et fréquemment à des arythmies (FA, flutter). La stratégie du contrôle du rythme doit être maintenue aussi longtemps que possible car les patients sont moins symptomatiques en rythme sinusal. Si la fibrillation atriale récidive malgré tout, un contrôle prudent de la fréquence ventriculaire est alors indiqué en favorisant les bêtabloquants comme traitement de premier choix. En effet, tant la digoxine que les anticalciques se lient aux fibrilles d’amyloïde et s’accumulent dans les tissus infiltrés, ce qui augmente le risque de toxicité médicamenteuse.15,16 Du moment où une arythmie atriale est diagnostiquée, il est très important de prescrire une anticoagulation orale car le risque d’AVC cardio-embolique est élevé, indépendamment du score CHA2DS2-Vasc.
L’infiltration des voies de conduction lors d’une amyloïdose peut également causer divers degrés de bloc atrio-ventriculaire (BAV). L’implantation d’un pacemaker est indiquée selon les mêmes recommandations que pour les BAV d’origine dégénérative. La prise en charge est cependant moins bien établie quand il s’agit de la prévention de la mort subite. Même si le décès sur troubles du rythme cardiaque semble fréquent dans cette population, il n’a pas pu être démontré qu’un défibrillateur en prévention primaire diminue ce risque.17
Traitements modifiant le pronostic
Le traitement de l’amyloïdose AL s’inscrit dans la gamme des traitements du myélome multiple et relève du spécialiste en oncohématologie. Ce traitement complexe, dont le principe est une chimiothérapie ± transplantation de moelle osseuse, ne sera pas abordé dans cet article.

Fig 7: Centres de formation de catégorie A pour la Cardiologie en Suisse
La seule option de traitement de l’AC ATTR a longtemps été la transplantation hépatique limitée à des jeunes patients atteints d’AC ATTRm. Mais ces dernières années, de nouvelles molécules ont montré leur efficacité, dont la première à être disponible en Suisse pour l’AC ATTR est le tafamidis (Vyndaquel®). Il s’agit d’un médicament qui stabilise la protéine TTR pour qu’elle conserve sa structure tétramérique et dont l’évidence thérapeutique repose sur une étude randomisée incluant >400 patients. Cette étude montre un important bénéfice au niveau de la mortalité (diminution absolue de 13%), des hospitalisations (diminution absolue de 32%) ainsi que de la qualité de vie à 30 mois de traitement.18 Le bénéfice semble être plus important chez des patients avec une maladie moins avancée se traduisant par un stade fonctionnel NYHA I–II. Sur la base de ces évidences le tafamidis a été mis sur la liste de spécialité de l’OFSP en décembre 2021, mais limitant le droit de prescription aux centres suisses de formation de Cardiologie de catégorie A (Fig. 7). Le remboursement du traitement est limité aux patients réunissant les critères suivants: diagnostic d’AC ATTR selon l’algorithme (Fig. 4), ≥1 épisode d’insuffisance cardiaque aiguë nécessitant des diurétiques, stade fonctionnel NYHA I–II, concentration de NT-proBNP >600pg/ml, distance de marche de 6 minutes >100mètres, espérance de vie de deux ans minimum.19
À noter que le médicament reste pour l’instant très onéreux, avec un prix de >10000frs/mois. De ce fait, il est extrêmement important de discuter l’indication du traitement entre le patient, le généraliste et le spécialiste avant toute demande de remboursement et dans certains cas avant même de suivre l’algorithme diagnostique.
Un autre groupe de traitements, appelé «TTR gene silencers» et dont le principe est d’interrompre la synthèse de TTR au niveau moléculaire, a été développé ces dernières années. Il existe actuellement deux molécules, le patisiran20 et l’inotersen,21 qui sont disponibles en Suisse et indiquées pour le traitement de la polyneuropathie en cas d’amyloïdose familiale (TTRm). Dans un sous-groupe prédéfini de patients avec atteinte cardiaque, il a été démontré que le patisiran permet d’obtenir une amélioration de la fonction systolique du VG et une diminution des valeurs de NT-proBNP.22 Par contre, il n’est pour l’instant pas remboursé pour l’AC . Des études de phase III sont actuellement en cours, et d’autres molécules viendront probablement élargir l’arsenal thérapeutique de l’AC ces prochaines années.
Littérature:
1 McDonagh T et al.: 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2021; 42: 3599-726 2 Hahn et al.: Endomyocardial biopsy characterization of heart failure with preserved ejection fraction and prevalence of cardiac amyloidosis. JACC Heart Fail 2020; 8: 712-24 3 Gonzalez-Lopez E et al.: Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36: 2585-94 4 Bennani-Smires Y et al.: Pilot study for left ventricular imaging phenotype of patients over 65 years old with heart failure and preserved ejection fraction: the high prevalence of amyloid cardiomyopathy. Int J Cardiovasc Imaging 2016; 32: 1403-13 5 Castano A et al.: Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 2017; 38: 2879-87 6 Longhi S et al.: Coexistence of degenerative aortic stenosis and wild-type transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging 2016; 9: 325-7 7 Condoluci A et al.: Management of transthyretin amyloidosis. Swiss Med Wkly 2021; 151: w30053 8 Fontana M et al. Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2015; 132: 1570-9 9 Banypersad SM et al.: Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: an equilibrium contrast cardiovascular magnetic resonance study. Circ Cardiovasc Imaging 2013; 6: 34-9 10 Dispenzieri A et al.: Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet 2010; 375: 1721-8 11 Gillmore JD et al.: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016; 133: 2404-12 12 Rapezzi C et al.: Systemic cardiac amyloidosis disease profile and clinical courses of the three main types. Circulation 2009; 120: 1203-12 13 Kumar S et al.: Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012; 30: 989-95 14 Buss SJ et al.: Longitudinal left ventricular function for prediction of survival in systemic light-chain amyloidosis: incremental value compared with clinical and biochemical markers. J Am Coll Cardiol 2012; 60: 1067-76 15 Muchtar E et al.: Digoxine use in systemic light chain amyloidosis – contra-indicated or cautious use? Amyloid 2018; 25: 86-92 16 Gertz M, Rodney H et al.: Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol 1985; 55: 1645 17 Lyn D et al.: Implantable cardioverter defibrillators in patients with cardiac amyloidosis. J Cardiovasc Electrophysiol 2013; 24: 793-8 18 Maurer MS et al.: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379: 1007-16 19 Liste des spécialités, Office fédéral de la santé publique OFSP, octobre 2022 20 Adams D et al.: Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 11-21 21 Benson MD et al.: Inotersen treatment for patient with hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 22-31 22 Solomon S et al.: Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation 2019; 139: 431-43 23 Hugelshofer S et al.: Cardiac amyloidosis: a rare disease? Rev Med Suisse. 2019; 15: 1054-9
Das könnte Sie auch interessieren:

BPCO: identification de patients non diagnostiqués
De nombreuses personnes atteintes de BPCO et/ou d’asthme n’ont jamais été diagnostiquées et ne sont donc pas traitées. Plusieurs études publiées ces derniers mois se penchent sur ce ...

Diarrhée chronique: déroulement de l’examen
La diarrhée chronique est le symptôme de différentes maladies. Le Pr Alain Schoepfer a expliqué comment rechercher au mieux le facteur déclenchant dans son exposé lors du congrès annuel ...

Réduction, voire arrêt des corticoïdes grâce à la biothérapie?
Les corticoïdes oraux constituent le traitement de référence des exacerbations de l’asthme et sont également utilisés pour celles de la BPCO. Cette norme s’appuie toutefois sur des ...