
Desserrer l’étau de la CMH
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
La cardiomyopathie hypertrophique (CMH) est définie par une hypertrophie ventriculaire gauche qui n’est causée par aucune autre pathologie.1 Elle est généralement plus fréquente qu’on ne le pense et touche environ 1 adulte sur 500.2 La détection précoce des signes de CMH et une évaluation ciblée sont essentielles, car il s’agit d’une maladie évolutive pouvant entraîner des complications sévères. Disponible depuis peu, le mavacamten, un inhibiteur spécifique et réversible de la myosine, est le premier traitement médicamenteux ciblé pour les patients atteints de CMH obstructive symptomatique, comme on a pu l’apprendre lors d’un symposium satellite organisé dans le cadre du congrès annuel de la SSC.
Le diagnostic de la CMH est souvent tardif, voire n’est jamais posé, car les symptômes ne sont pas spécifiques. Une angine de poitrine, une dyspnée, des palpitations, une syncope, un épuisement, une intolérance à l’effort, qui peuvent se manifester à des degrés très divers, sont des symptômes qui doivent faire penser à une CMH.1 Les antécédents familiaux, un ECG pathologique et un souffle systolique peuvent également indiquer une CMH. L’identification de ces indicateurs potentiels de CMH est une première étape importante dans le diagnostic de cette maladie évolutive qui peut entraîner une insuffisance cardiaque évolutive, une fibrillation auriculaire, un accident vasculaire cérébral ainsi qu’une mort cardiaque subite.2
Diagnostic efficace de la CMH: évaluation de l’obstruction au moyen de l’échographie de stress physiologique
Morphologiquement, la CMH se traduit par une hypertrophie asymétrique du ventricule gauche, qui ne peut pas être expliquée uniquement par l’effort. La valeur limite pour le diagnostic de la CMH est une épaisseur de paroi ≥15mm (≥13mm en cas d’antécédents familiaux positifs) au niveau d’au moins un segment myocardique.1 Chez environ 70% des patients atteints de CMH, l’hypertrophie croissante du septum entraîne une obstruction de la chambre de chasse du ventricule gauche (LVOTO).3 Comme la cardiomyopathie hypertrophique obstructive (CMHO) est associée à un pronostic moins favorable que la CMH non obstructive, il est important de rechercher activement une obstruction lors du diagnostic.4 «L’obstruction produit un souffle cardiaque méso- ou télésystolique, qui rappelle un peu le rétrécissement aortique et qui est généralement entendu au niveau du foyer d’Erb, plus rarement au-dessus de l’aorte. Une caractéristique importante est que le souffle systolique est dynamique dans le cas de la CMHO. Il peut être renforcé par une provocation au moyen d’une manœuvre de Valsalva ou en se levant de la position accroupie»1, a expliqué le PD Dr méd. Pierre Monney du CHUV à Lausanne. Chez la majorité des patients atteints de CMHO, l’ECG montre des anomalies telles qu’un voltage élevé, une onde T et un segment ST anormaux ainsi que des ondes Q pathologiques (ondes Q de pseudo-nécrose) dans différentes combinaisons.1
L’échocardiographie est essentielle pour le diagnostic et le suivi de la CMH. Outre la mesure de l’épaisseur de la paroi, il convient de rechercher des indicateurs d’une LVOTO, tels qu’un mouvement systolique antérieur («systolic anterior motion», SAM) de la valve mitrale, des anomalies des muscles papillaires et une augmentation du gradient de la LVOTO (≥ 30 mmHg). «Il est important de noter que la LVOTO est indétectable au repos chez environ la moitié des patients atteints de CMHO. Il s’agit ici d’une obstruction latente qui doit être provoquée par une manœuvre de Valsalva ou un effort physique»,3 a souligné le PDDrméd. Monney. «L’échocardiographie de stress physiologique avec mesure du gradient de la LVOTO immédiatement après un effort sur vélo ergomètre ou tapis roulant est la méthode la plus efficace pour détecter une LVOTO latente.1 Le tapis roulant paraît y provoquer les gradients LVOT les plus élevés5»,a-t-il ajouté.
L’imagerie par résonance magnétique (IRM) cardiaque sert à l’évaluation morphologique chez les patients dont les résultats de l’échocardiographie ne sont pas clairs, ainsi qu’à la caractérisation des tissus et à la quantification de la fibrose («late gadolinium enhancement»). Elle est également utile pour exclure d’importants diagnostics différentiels tels que la maladie de Fabry, l’amylose ou d’autres phénocopies.6
Nouveau traitement ciblé de la CMH
Sur le plan physiopathologique, la CMH repose sur un dysfonctionnement des sarcomères myocardiques. Les sarcomères, qui sont la plus petite unité contractile des cellules musculaires, assurent une contraction et une relaxation régulières. Les liaisons transversales actine-myosine sont déterminantes pour la contractilité. En cas de myocarde sain, 40 à 50% des têtes de myosine sont inactives (état «off»). Cependant, seuls 15 à 20% environ des têtes de myosine sont inactives en cas de CMH, ce qui entraîne un excès de liaisons transversales actine-myosine (Fig. 1).7–11 Les conséquences de ce dysfonctionnement sont notamment une hypercontractilité accompagnée d’une faible efficacité mécanique, d’une diminution de la relaxation et d’une hypertrophie.
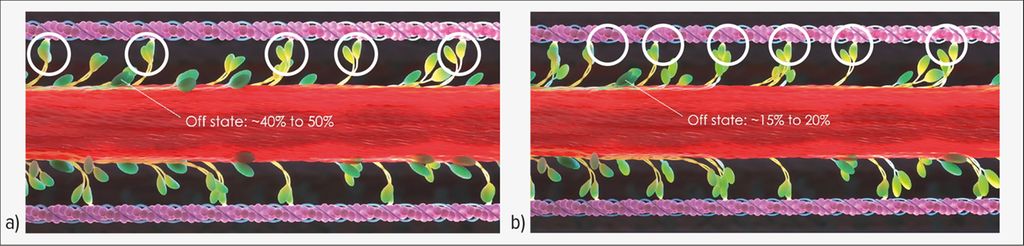
Fig. 1: Dysfonctionnement des sarcomères dans la cardiomyopathie hypertrophique (CMH). a) En cas de myocarde sain, 40 à 50% des têtes de myosine sont inactives et ne forment pas de liaisons transversales avec l’actine. b) En cas de CMH, seuls 15 à 20% des têtes de myosine sont inactives, ce qui entraîne un excès de liaisons transversales actine-myosine
Avec le mavacamten (Camzyos®), on dispose pour la première fois d’un médicament qui s’attaque à ce mécanisme pathologique. Il se lie de manière réversible à la myosine cardiaque, réduisant ainsi le nombre de liaisons transversales actine-myosine. «Cela entraîne une réduction de l’hypercontractilité, une amélioration de la relaxation, une diminution de la rigidité du tissu cardiaque ainsi qu’une amélioration de l’énergie et de la compliance du muscle cardiaque»,7–11 a expliqué la PDDre méd. Annina Vischer de l’Hôpital universitaire de Bâle. En Suisse, le mavacamten est autorisé depuis avril 2023 pour améliorer les performances physiques et les symptômes associés chez les patients adultes atteints de CMHO symptomatique (classe II–III de la NYHA).12
Il a notamment été étudié dans le cadre de deux études de phase III, EXPLORER-HCM13 et VALOR-HCM14. Elles ont inclus des patients adultes symptomatiques (classe II ou III de la NYHA dans EXPLORER-HCM ou III–IV dans VALOR-HCM) présentant une CMHO documentée et un gradient maximal de la LVOTO ≥50mmHg au repos ou après provocation). Dans l’étude VALOR-HCM, les patients devaient répondre aux critères de la thérapie de réduction septale (SRT). Dans les deux études, le mavacamten a été comparé à un placebo (+/- traitement de fond).
Dans l’étude EXPLORER-HCM, après 30 semaines, plus du double des patients dans le groupe sous traitement actif par rapport au groupe sous placebo (37% vs 17%; IC à 95%: 8–30,1; p=0,0005) ont atteint le critère d’évaluation primaire – défini comme une augmentation de la consommation maximale d’oxygène (VO2peak) de ≥1,5ml/kg/min plus une amélioration de ≥1classe de la NYHA ou une augmentation de la VO2peak de ≥3ml/kg/min sans détérioration de la classe de la NYHA.13 «La réduction de 48mmHg du gradient de la LVOTO sous mavacamten, alors qu’il est resté inchangé sous placebo, est également très impressionnante»,13 a déclaré la PDDre méd. Vischer. «Un tableau clinique similaire a été observé pour les biomarqueurs: sous mavacamten, on a constaté une diminution significative et parfois une normalisation du NT-proBNP ainsi que de la hs-cTnI. Cela indique que le traitement entraîne des changements profonds au niveau cardiaque.»13
L’étude VALOR-HCM a évalué si les patients remplissaient toujours les critères de la SRT après 16 semaines de traitement. Il s’est avéré que ce n’était plus le cas pour 82% des patients sous mavacamten (vs 23%; IC à 95%: 43,99–73,87; p<0,0001).14 Les résultats concernant les critères d’évaluation secondaires (diminution du gradient de la LVOTO et des biomarqueurs) étaient comparables à ceux obtenus dans l’étude EXPLORER-HCM.14 Malgré son effet inotrope négatif, les deux études ont également montré que l’influence du mavacamten sur la fraction d’éjection ventriculaire gauche (FEVG) était très faible et n’était pas significativement plus importante que sous placebo.13,14
Le traitement par le mavacamten est généralement débuté à une dose de 5mg/j. La dose est titrée dans les semaines suivantes sur la base du gradient de la LVOTO et de la FEVG (dose maximale: 15mg/j).12 Le mavacamten est principalement métabolisé par les enzymes du cytochrome P450, il faut donc faire attention aux interactions possibles.12
Conclusion
Le diagnostic de la CMH commence par l’identification des indicateurs de la CMH et est confirmé par l’ECG ainsi que l’imagerie multimodale. Une attention particulière est accordée à la recherche active d’une obstruction latente au moyen de l’échocardiographie de stress physiologique. Disponible depuis peu, l’inhibiteur de la myosine mavacamten (Camzyos®) est le premier traitement médicamenteux spécifique et ciblé qui s’est avéré efficace dans des études de phase III chez les patients atteints de CMHO symptomatique.
Source:
Symposium de l’entreprise Bristol-Myers Squibb SA dans le cadre du congrès annuel de la SSC, du 21 au 23 juin 2023, Bâle
Littérature:
1 Elliott PM et al.: 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35: 2733-79 2 Maron BJ et al.: Diagnosis and evaluation of hypertrophic cardiomyopathy: JACC State-of-the-Art review. J Am Coll Cardiol 2022; 79: 372-89 3 Maron MS et al.: Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114: 2232-9 4 Maron MS et al.: Contemporary natural history and management of nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2016; 67: 1399-409 5 Reant P et al.: Upright treadmill vs. semi-supine bicycle exercise echocardiography to provoke obstruction in symptomatic hypertrophic cardiomyopathy: a pilot study. Eur Heart J Cardiovasc Imaging 2018; 19: 31-8 6 Rapezzi C et al.: Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013; 34: 1448-58 7 Anderson RL et al.: Deciphering the super relaxed state of human β-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc Natl Acad Sci USA 2018; 115: E8143-52 8 Nag S et al.: The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat Struct Mol Biol 2017; 24: 525-33 9 Spudich JA et al.: Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflugers Arch 2019; 471: 701-17 10 Alamo L et al.: Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. eLife 2017; 6: e24634 11 Sequeira V et al.: Energetic drain driving hypertrophic cardiomyopathy. FEBS Lett 2019; 593: 1616-26 12 Information professionnelle de mavacamten: swissmedicinfo.ch 13 Olivotto I et al.: Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020; 396: 759-69 14 Desai MY et al.: Myosin inhibition in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy. J Am Coll Cardiol 2022; 80: 95-108
Bristol-Myers Squibb SA fournit les références listées sur demande.
Information professionnelle abrégée
▼ Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave. Voir la rubrique «Effets indésirables» pour les modalités de déclaration des effets secondaires.
CAMZYOS® (mavacamten). MGI: Peut provoquer une insuffisance cardiaque due à un dysfonctionnement systolique. Examens échocardiographiques de la FEVG nécessaire avant et pendant l’utilisation. Début du traitement avec une FEVG <55% non recommande. Interruption du traitement si FEVG <50% ou en cas de dégradation de l’état clinique. Prudence lors de l’administration concomitante de certains inhibiteurs et inducteurs du CYP450 en raison du risque d’insuffisance cardiaque. I:Traitement des patients adultes présentant une cardiomyopathie hypertrophique obstructive (CMHo) symptomatique (NYHA II-III) afin d’améliorer la performance physique et les symptômes associes. P: 2,5 mg, 5 mg, 10 mg ou 15 mg 1x/j. La posologie initiale recommandée est de 5 mg 1x/j, la dose quotidienne maximale est de 15 mg 1x/j. L’instauration ou l’augmentation de dose de Camzyos chez les patients avec une FEVG < 55 % ne sont pas recommandées. Une évaluation régulière de la FEVG et du gradient de la CCVG par Valsalva est nécessaire pour une titration méticuleuse afin d’atteindre un gradient-cible de CCVG par Valsalva adéquat et de maintenir une FEVG ≥50%. Pour les instructions de titration pendant les phases d’instauration et de maintien, la modification de la posologie avec des médicaments concomitants et l’arrêt du traitement, voir l’information professionnelle complète. En cas de FEVG < 50 % pendant la prise de Camzyos, le traitement doit être interrompu. CI: Hypersensibilité au principe active ou aux excipients. Grossesse. MG&P: Mavacamten réduit la FEVG et peut déclencher l’insuffisance cardiaque par dysfonctionnement systolique. La posologie doit tenir compte d’une réduction asymptomatique de la FEVG, des affections intercurrentes et des arythmies. Evaluation régulière de l’état clinique avant et pendant le traitement par CAMZYOS. IA: Un ajustement posologique du mavacamten et/ou un contrôle clinique supplémentaire sont recommandes en cas d’une instauration, arrêt ou ajustement posologique d’un traitement concomitant avec un inhibiteur puissant du CYP2C19 et doivent être envisager avec inhibiteur modéré ou faible du CYP2C19 ou puissant du CYP3A4. En cas d’arrêt ou de diminution posologique du traitement concomitant par un inducteur puissant du CYP2C19 ou du CYP3A4, des évaluations cliniques supplémentaires sont recommandées. En cas d’instauration ou d’augmentation posologique d’un inducteur puissant, une adaptation posologique du mavacamten et/ou des évaluations cliniques doivent être envisagées. L’administration intermittente d’inhibiteurs ou d’inducteurs puissants du CYP2C19 ou d’inhibiteurs puissants ou d’inducteurs puissants du CYP3A4 n’est pas recommandée. EI: Vertiges, insuffisance cardiaque, dysfonctionnement systolique (FEVG < 50 %). P: Gélules de 2,5 mg, 5 mg, 10 mg ou 15 mg: 28. (B). TA: Bristol-Myers Squibb SA, Hinterbergstrasse 16, 6312 Steinhausen. Information professionnelle complète sur www.swissmedicinfo.ch. (V001)
Avec l’aimable soutien de Bristol-Myers Squibb SA
CV-CH-2300046 07.2023