Kardiale Transthyretin-Amyloidose in der Erstvorstellung
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Bereits die Erstvorstellung kann wichtige Hinweise für das Vorliegen einer kardialen Transthyretin(ATTR)-Amyloidose liefern. Die im Vergleich zur Vergangenheit einfache Diagnosesicherung durch spezialisierte Zentren ohne Notwendigkeit einer routinemäßigen Herzmuskelbiopsie in Kombination mit zur Verfügung stehenden Therapeutika macht eine frühe Diagnose immer wichtiger. Dennoch ist die kardiale Amyloidose bis heute stark unterdiagnostiziert.1, 2
Keypoints
-
Die kardiale ATTR-Amyloidose ist eine unterdiagnostizierte, therapierbare Erkrankung.
-
Bereits der frühe Patient:innenkontakt liefert erste Hinweise auf das Vorliegen der Erkrankung.
-
Klinisch und anamnestisch können sich Symptome bzw. Zeichen einer Herzinsuffizienz, peripherer Polyneuropathien, eines beidseitigen Karpaltunnelsyndroms sowie lumbaler Spinalkanalstenosen zeigen.
-
Elektrokardiografisch herrscht im Gegensatz zur Bildgebung häufig eine Diskrepanz hinsichtlich Hypertrophiezeichen, Pseudoinfarktmustern, Vorhofflimmern sowie Reizleitungsstörungen (AV-/Schenkelblock).
-
Wichtige echokardiografische Hinweise sind eine nicht durch eine erhöhte Nachlast erklärbare, linksventrikuläre Wandverdickung, vergrößerte Vorhöfe und milde Perikardergüsse.
Die Amyloidose beschreibt eine Krankheit, bei der es zur extrazellulären, infiltrativen Ablagerung von abnormen Proteinen im Interstitium kommt. Diese andersartigen Proteine lagern sich in Form von unlöslichen Beta-Fibrillen ab und können lokalisiert oder systemisch auftreten. An ihrem Ablagerungsort verursachen sie eine Organdysfunktion. Es sind 42 Vorläuferproteine bekannt, die sich zu unlöslichen Fibrillen anhäufen.3 Einige davon weisen eine hohe kardiale Affinität auf. Die häufigsten kardialen Amyloidosen sind die ATTR- und die Leichtketten(AL)-Amyloidose.
Das Transportprotein Transthyretin (TTR) wird zu 95% in der Leber und zu kleinen Teilen unter anderem im Plexus choroideus und im Netzhautpigmentepithel gebildet.4 Es gibt zwei ATTR-Amyloidose-Formen: die hereditäre Form ATTRv („variant“) oder auch hATTR; die senile Form ATTRwt („wild-type“). Bei der vATTR-Amyloidose kommt es durch Punktmutationen im TTR-Gen zu einer fehlgebildeten Variante, die sich je nach Mutationsart unterschiedlich manifestiert, jedoch meist das Herz, das periphere und autonome Nervensystem und den GI-Trakt betrifft. Es handelt sich um einen autosomal-dominanten Erbgang.
Die erworbene Form stellt die wtATTR-Variante dar, bei welcher es zu einer Fehlfaltung des normalen TTR kommt. Sie betrifft primär das Herz und das periphere Nervensystem und tritt vor allem im höheren Lebensalter auf (Männer ≥ 65a, Frauen ≥ 70a).
Anamnese & körperliche Untersuchung
Erste Hinweise auf das Vorliegen einer ATTR-Amyloidose ergeben sich schon im frühen Patient:innenkontakt, wie auch im folgenden Fallbeispiel eines 80 Jahre alten Patienten (Herr AB). Dieser stellt sich auf Zuweisung aus dem niedergelassenen Bereich aufgrund einer linksventrikulären Hypertrophie und eines „speckled myocardium“ in der kardiologischen Ambulanz vor. Es handelt sich um einen sehr sportlichen Patienten, der bis vor Kurzem noch 130 Watt am Heimtrainer fahren konnte, nun jedoch eine Belastungsdyspnoe im Sinne einer „New York Heart Association“(NYHA)-Klassifikation von II entwickelt hat. Als kardiale Vorgeschichte liegt eine Zweigefäßerkrankung mit perkutaner Intervention der rechten und linken Koronararterie drei Jahre zurück. Vor einigen Jahren wurde eine Karpaldachspaltung aufgrund eines beidseitigen Karpaltunnelsyndroms durchgeführt. Außerdem besteht der Zustand nach Nephrektomie aufgrund eines Traumas in jungen Jahren sowie eine Prostatektomie bei einem N. prostatae.
Ein typischer Patient mit einer ATTR-Kardiomyopathie (CM) ist männlich, über 65 Jahre alt und unterliegt der „Wild-type“-Form der ATTR-Amyloidose. Die hereditäre Variante ist in Bezug auf das Geschlecht ausgeglichen und abhängig von der zugrundeliegenden Mutation in ihrer klinischen Manifestation weitaus heterogener. Das Hauptaugenmerk in der folgenden Beschreibung der klinischen Präsentation liegt hier auf der häufigeren Variante, der wtATTR-Amyloidose. Aufgrund der extrazellulären kardialen Ablagerung der unlöslichen Amyloidfibrillen kommt es im Verlauf zu einer restriktiven Kardiomyopathie, die sich klinisch in Herzinsuffizienzsymptomen und -zeichen äußert. Extrakardiale Manifestationen sind zumeist neurogener Natur und betreffen das periphere und autonome Nervensystem. Das häufigste klinische extrakardiale Zeichen ist das (bilaterale) Karpaltunnelsyndrom, das bereits sehr früh und vor kardialer Manifestation beobachtet werden kann.5 Weiters kann es zu einer peripheren, bilateralen sensomotorischen Polyneuropathie, beginnend in den unteren Extremitäten, kommen.6 Dysautonomien zeigen sich in Form von orthostatischen Hypotonien, erektiler Dysfunktion und wechselndem Stuhlverhalten.7 Weitere Hinweise auf das Vorliegen einer kardialen ATTR-Amyloidose sind lumbale Spinalkanalstenosen und atraumatische Bizepssehnenrupturen.8,9 Ebenfalls wird häufig über Schwindel oder (Prä-)Synkopen berichtet, welche auf eine orthostatische Hypotonie oder Herzrhythmusstörungen zurückzuführen sind.Eine weitere Ursache können eine gewisse Intoleranz und Therapierefraktärität gegenüber kardiologischen Standardtherapeutika in Form von symptomatischer Hypotonie und Bradykardie sein.10,11 Dies betrifft vor allem Betablocker.
Laborchemische Untersuchung
Der Stellenwert der Laboruntersuchung im Erstkontakt bzw. Screening einer kardialen ATTR-Amyloidose ist eher untergeordnet und erst im weiteren Verlauf zur Risikostratifizierung, Therapieüberwachung oder Differenzialdiagnostik anderer Formen der Amyloidose (v.a. der AL-Amyloidose) durch spezialisierte Zentren relevant.12 Der Ein-/Ausschluss einer AL-Amyloidose ist jedoch aufgrund der Notwendigkeit der frühen Therapieinitiierung bei AL-Amyloidose sehr zeitkritisch zu betrachten. Für den Erstkontakt mit Patient:innen mit Verdacht auf ATTR-Kardiomyopathie kann zusammenfassend grob festgehalten werden, dass sie disproportional zum Grad der bestehenden Herzinsuffizienz erhöhte natriuretische Peptide (NT-proBNP) und konstant, leicht erhöhte Troponine aufweisen.
12-Kanal-Elektrokardiogramm
Das Elektrokardiogramm (EKG) kann wichtige Hinweise auf das Vorliegen einer ATTR-CM liefern. Bei Erstvorstellung zeigte sich im 12-Kanal-EKG von Herrn AB unter anderem ein normofrequentes Vorhofflimmern, eine maximale QRS-Amplitude in den Extremitätenableitungen von 0,6mV und Fehlen von Hypertrophiezeichen bei bestehender Hypertrophie in der Bildgebung (Abb. 1a). Weiters zeigt sich eine verzögerte R-Progression über V1–V3. In Zusammenschau ist dieser Befund suspekt für das Vorliegen einer kardialen Amyloidose. So hat eine Arbeit von Schrutka et al. einen EKG-Algorithmus mittels maschinellen Lernens entwickelt, um den klinischen Verdacht einer kardialen Amyloidose zu erhärten(Abb. 1b).13Im vorliegenden Fallbeispiel wären die Algorithmuskriterien des 1. Musters erfüllt und ein weiteres diagnostisches Prozedere ist empfohlen.
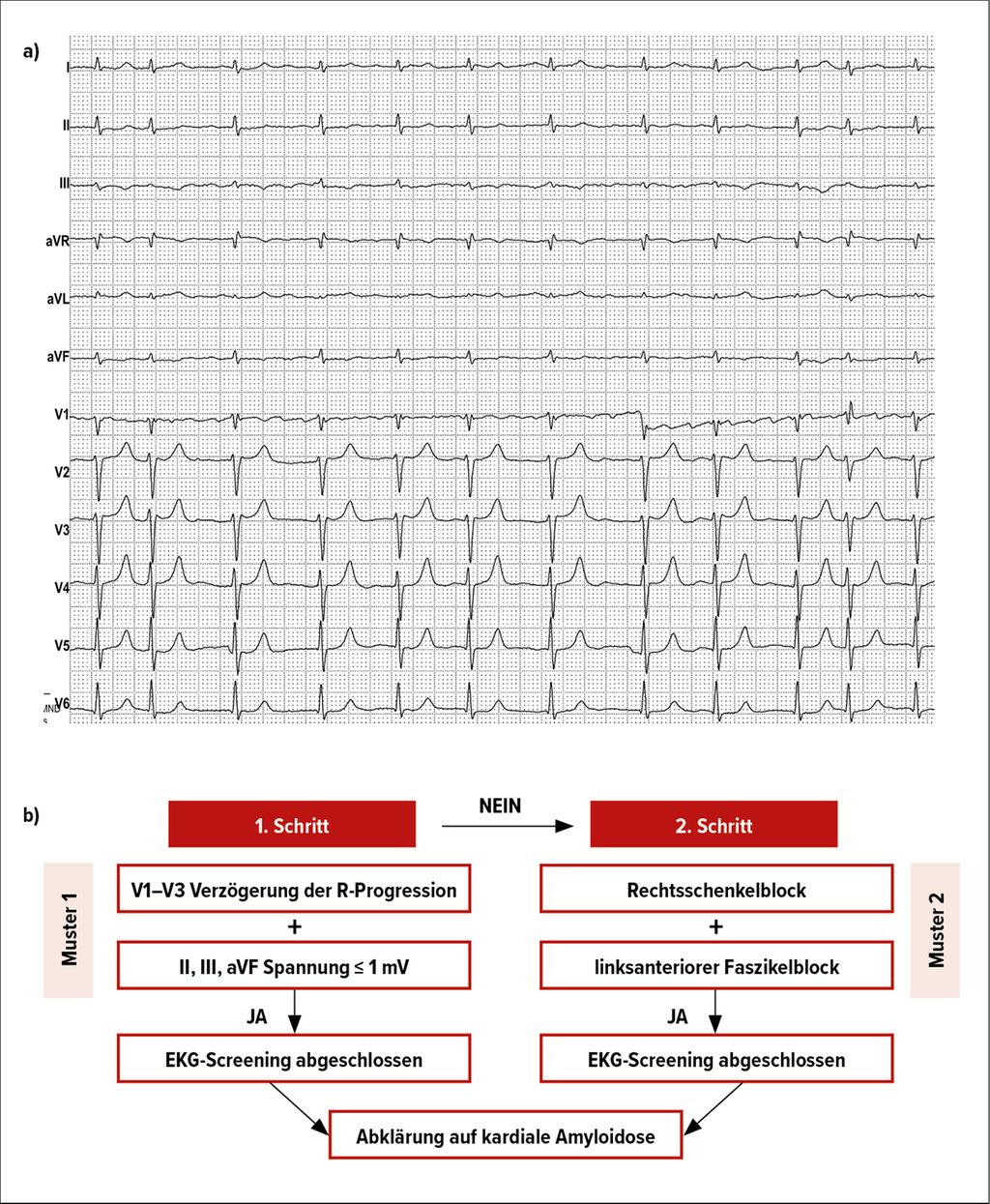
Abb. 1: a) EKG bei Erstvorstellung; b) EKG-Algorithmus für Amyloidosescreening (nach Schrutka L et al. 2022)13
Patient:innen mit einer kardialen Amyloidose präsentieren sich häufig mit einem hypertrophen Phänotyp in der Bildgebung, im Gegensatz dazu zeigt sich jedoch selten ein korrelierendes Hypertrophiezeichen im EKG.14 Diese Diskrepanz kann in fortgeschrittenen Stadien so weit gehen, dass bei Vorliegen einer linksventrikulären Hypertrophie eine Niedervoltage herrscht. Die Hypertrophie bei der kardialen Amyloidose ist nicht auf die Kardiomyozyten, sondern auf die Ablagerung der pathologischen Betafibrillen im Extrazellularraum zurückzuführen. Weitere zugrundeliegende Pathomechanismen für diese Diskrepanz sind in Diskussion.15 Ein Pseudoinfarktmuster, definiert als pathologische Q-Zacken in zumindest zwei konkordanten Ableitungen ohne eine dazupassende koronare Herzkrankheit, wird häufig beobachtet.16 Weiters hat Vorhofflimmern sehr hohe Prävalenz in der ATTR-CM.17 Dies ist vor allem auf die Amyloidablagerungen und auf die linksatriale Dilatation zurückzuführen. Außerdem kommt es häufig zu atrioventrikulären und Schenkelblockierungen, welche bis zur Schrittmacherabhängigkeit führen können.18
Transthorakale Echokardiografie
Eine ATTR-CM führt durch die interstitiellen Amyloidablagerungen zu einem hypertrophen Phänotyp in der transthorakalen Echokardiografie. Die Prävalenz bei Patient:innen mit einer Herzinsuffizienz mit erhaltener Pumpfunktion und bei Patient:innen mit einer („low-flow/low-gradient“) Aortenstenose ist hoch.19,20 Wichtige morphologische Merkmale sind kleine oder normal dimensionierte, jedoch wandverdickte Ventrikel, vergrößerte Vorhöfe und minimale Perikardergüsse, vor allem im Bereich des rechten Vorhofes (Abb. 2).
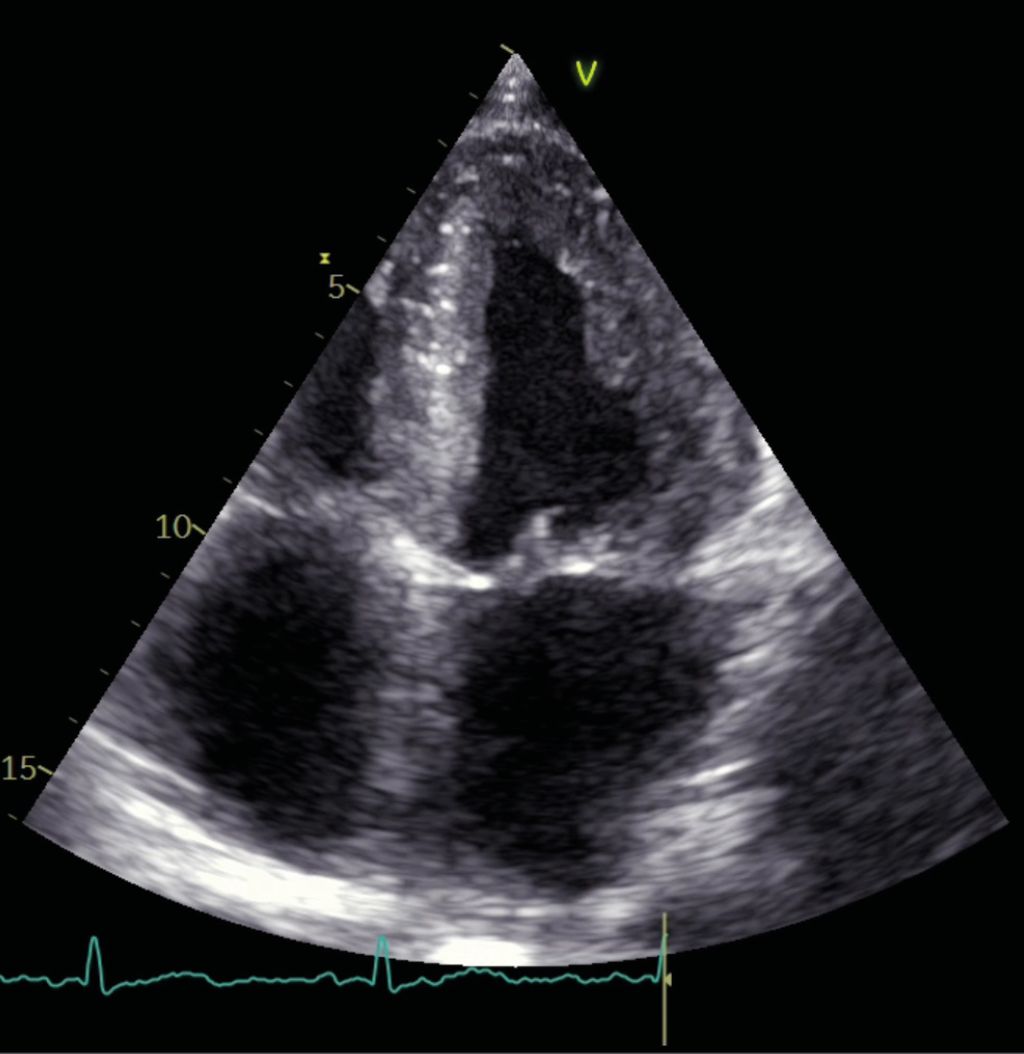
Abb. 2: Apikaler 4-Kammer-Blick bei transthorakaler Echokardiografie bei Herrn AB
Herr AB zeigte bei Erstvorstellung in der transthorakalen Echokardiografie eine erhaltene Pumpfunktion mit einer diastolischen Dysfunktion, einer Septumdicke von 16mm sowie dilatierte Vorhöfe. Die Verdickung in der Bildgebung betrifft die linksventrikuläre und rechtsventrikuläre Wand, das interatriale Septum als auch den Klappenapparat.21 Aufgrund der guten radialen linksventrikulären Funktion ist die Pumpfunktion häufig erhalten, die diastolische Funktion aufgrund der restriktiven Komponente jedoch eingeschränkt. Die diastolische Dysfunktion zeigt sich häufig bereits vor den Wandverdickungen. Eine wichtige ergänzende Modalität in der transthorakalen Echokardiografie ist die Messung der longitudinalen linksventrikulären Funktion im „strain imaging“. Dieses zeigt bei der ATTR-CM typischerweise eine „Strain“-Reduktion mit relativer apikaler Aussparung, dem „apical sparing“ (gute Funktion der apikalen Segmente, weniger Wandstress).22,23 Dies präsentiert sich im „bull’s-eye plot“ wie ein „Cherry on the top“-Muster und kann vor allem in der Differenzialdiagnostik der Myokardwandverdickungen behilflich sein (Abb. 3).
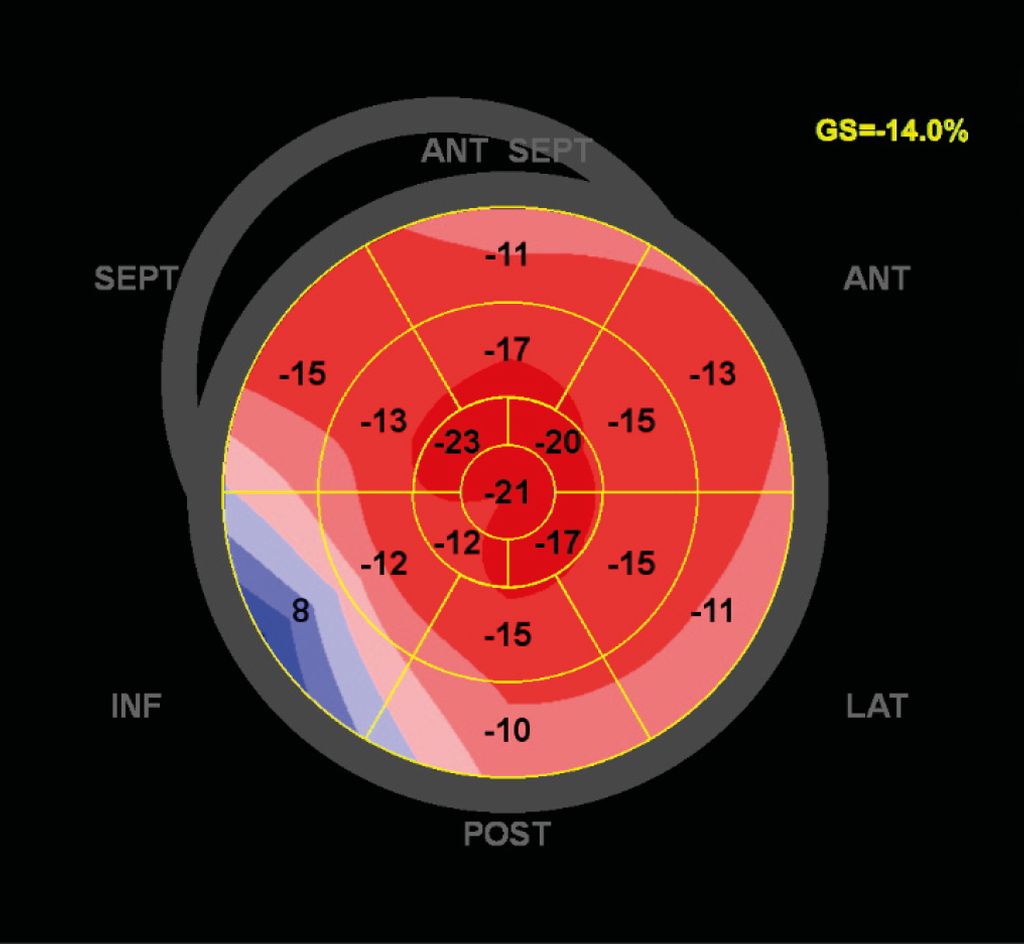
Abb. 3: „Strain imaging bull’s eye plots“ von Herrn AB mit „apical sparing“
Verdacht auf kardiale (ATTR-)Amyloidose
Der Verdacht auf das Vorliegen einer kardialen (ATTR-)Amyloidose sollteimmer in Zusammenschau der Befunde gestellt werden. Befundkonstellationen ergeben sich aus der Anamnese, der körperlichen Untersuchung, dem EKG und der transthorakalen Echokardiografie. Besonders Patient:innen mit einem hypertrophen Phänotyp in der kardialen Bildgebung, definiert als linksventrikuläre Myokarddicke ≥14mm, die nicht durch eine erhöhte Nachlast erklärbar ist (arterielle Hypertonie, Aortenklappenstenose, u.a.) und in Verbindung mit anderen o.g. Hinweisen steht, sollten weiter abgeklärt werden. Patient:innengruppen mit einer hohen Prävalenz an ATTR-CM sind Patient:innen in einem fortgeschrittenen Lebensalter, mit einer Herzinsuffizienz mit erhaltener Pumpfunktion und mit einer „Low-flow/low-gradient“-Aortenklappenstenose.24
Besteht der Verdacht auf eine kardiale Amyloidose, sollte eine zeitnahe Überweisung an ein spezialisiertes Zentrum zur weiteren Diagnostik und gegebenenfalls Therapieeinleitung erfolgen. Bei Herrn AB bestätigte sich der Verdacht einer kardialen Amyloidose und nach laborchemischer Analyse einer kardialen Magnetresonanztomografie sowie einer Knochenszintigrafie wurde die Diagnose einer wtATTR-Amyloidose gestellt. Er stehtunter medikamentöser Therapie mit Tafamidis, erscheint regelmäßig zu Kontrolluntersuchungen und befindet sich stabil im NYHA-Stadium I mit regelmäßiger sportlicher Aktivität.
Literatur:
1 Oerlemans MIFJ et al.: Cardiac amyloidosis: the need for early diagnosis. Neth Heart J 2019; 27(11): 525-36 2 Manolis AS et al.: Cardiac amyloidosis: an underdiagnosed/underappreciated disease. Eur J Inern Med 2019; 67: 1-13 3 Cohen SIA et al.: Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 2013; 110(24): 9758-63 4 Sanguinetti C et al.: The journey of human transthyretin: synthesis, structure stability, and catabolism. Biomedicines 2022; 10(8): 1906 5 Milandri A et al.: Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail 2020; 22(3): 507-15 6 Adams D et al.: Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol 2021; 268(6): 2109-22 7 Barroso FA et al.: Characteristics of patients with autonomic dysfunction in the transthyretin amyloidosis outcomes survey (THAOS). Amyloid 2022; 29(3): 175-83 8 Godara A et al.: Association between spinal stenosis and wild-type ATTR amyloidosis. Amyloid 2021; 28(4): 226-33 9 Geller H I et al.: Association between ruptured distal biceps tendon and wild-type transthyretin cardiac amyloidosis. JAMA 2017; 318(10): 962-3 10 Ramsell S et al.: Beta-adrenergic antagonist tolerance in amyloid cardiomyopathy. Front Cardiovasc Med 2022; 9: 907597 11 Aimo A et al.: Safety and tolerability of neurohormonal antagonism in cardiac amyloidosis. Eur J Intern Med 2020; 80: 66-72 12 Castiglione V et al.: Use of biomarkers to diagnose and manage cardiac amyloidosis. Eur J Heart Fail 2021; 23(2): 217-30 13 Schrutka L et al.: Machine learning-derived electrocardiographic algorithm for the detection of cardiac amyloidosis. Heart 2022; 108(14): 1137-47 14 Cheng Z et al.: Utility of combined indexes of electrocardiography and echocardiography in the diagnosis of biopsy proven primary cardiac amyloidosis. Ann Noninvasive Electrocardiol 2011; 16(1): 25-9 15 Cipriani A et al.: Low QRS voltages in cardiac amyloidosis: clinical correlates and prognostic value. JACC CardioOncol 2022; 4(4): 458-70 16 Cheng Z et al.: The findings of electrocardiography in patients with cardiac amyloidosis. Ann Noninvasive Electrocardiol 2013; 18(2): 157-62 17 Donnellan E et al.: Atrial fibrillation in transthyretin cardiac amyloidosis: Predictors, prevalence, and efficacy of rhythm control strategies. JACC Clin Electrophysiol 2020; 6(9): 1118-27 18 Donnellan E et al.: Prevalence, incidence, and impact on mortality of conduction system disease in transthyretin cardiac amyloidosis. Am J Cardiol 2020; 128: 140-6 19 González-López E et al.: Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36(38): 2585-94 20 Ternacle J et al.: Aortic stenosis and cardiac amyloidosis: JACC review topic of the week. J Am Coll Cardiol 2019; 74(21): 2638-51 21 Aimo A et al.: Valve disease in cardiac amyloidosis: an echocardiographic score. Int J Cardiovasc Imaging 2023; 39(10): 1873-87 22 Phelan D et al.: Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 2012; 98(19): 1442-8 23 Marcoux J et al.: A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med 2015; 7(10): 1337-49 24 Bonderman D et al.: Diagnosis and treatment of cardiac amyloidosis: an interdisciplinary consensus statement. Wien Klin Wochenschr 2020; 132(23): 742-61
Das könnte Sie auch interessieren:

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

Inclisiran bei Patienten mit Statinintoleranz wirksam und sicher
Eine Analyse statinintoleranter Patienten aus dem Phase III Studienprogramm ORION zeigt, dass Inclisiran die LDL-Cholesterinspiegel kardiovaskulärer Hochrisikopatienten, die kein Statin ...