
Amyloidose als Ursache von Herzinsuffizienz mit erhaltener EF
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
In den letzten Jahren haben im Bereich der kardialen Amyloidose drei Paradigmenwechsel stattgefunden: 1) Die häufigste Form der Amyloidose (Transthyretin-Amyloidose) lässt sich mithilfe eines hauptsächlich auf bildgebenden Verfahren und nicht auf Biopsien beruhenden Ansatzes diagnostizieren; 2) es hat sich gezeigt, dass ein erheblicher Anteil der Patienten mit Herzinsuffizienz mit erhaltener EF an einer kardialen Amyloidose leidet; 3) eine krankheitsmodifizierende Therapie wurde zugelassen. Durch eine gute Zusammenarbeit zwischen Allgemeinmediziner, Kardiologe und in manchen Fällen Hämatologe können die von dieser tödlichen Krankheit betroffenen Patienten identifiziert, charakterisiert und behandelt werden.
Keypoints
-
Die kardiale Amyloidose (CA) vom ATTRwt-Typ ist keine Seltenheit und muss im Fall einer Herzinsuffizienz mit erhaltener Ejektionsfraktion in Betracht gezogen werden.
-
Nach formellem Ausschluss einer monoklonalen Gammopathie kann die ATTR-CA mittels 99mTc-DPD-Skelettszintigrafie/SPECT diagnostiziert werden.
-
Neue Medikamente gegen die ATTR-CA verlangsamen die Krankheitsprogression.
Die Prävalenz der Herzinsuffizienz nimmt stetig zu. Schätzungen zufolge liegt die Zahl der betroffenen Patienten in der Schweiz aktuell bei 150000, wobei knapp die Hälfte an einer Herzinsuffizienz mit erhaltener Ejektionsfraktion (HFpEF) leidet. Definiert ist diese durch eine LVEF >50%, das Vorliegen von Symptomen oder Zeichen einer Herzinsuffizienz und den Nachweis kardialer Anomalien, die mit einer diastolischen Dysfunktion/Erhöhung des linksventrikulären Füllungsdrucks vereinbar sind.1 Der erhöhte Füllungsdruck wird anhand echokardiografischer Merkmale, mithilfe invasiver Messungen oder durch einen Anstieg der natriuretischen Peptide (BNP/NT-proBNP) im Blut nachgewiesen. Die Patienten mit HFpEF stellen eine heterogene Population dar, die häufig ein fortgeschrittenes Alter und multiple Begleiterkrankungen aufweist, was die Identifikation der genauen Ätiologie erschwert. Als mögliche Ursachen kommen eine koronare Herzkrankheit, arterielle Hypertonie, Diabetes oder Adipositas infrage, wobei häufig eine Kombination mehrerer dieser Erkrankungen verantwortlich ist. Die Prognose bei dieser Form der Herzinsuffizienz ist ungünstig, und die zur Verfügung stehenden medikamentösen Therapien bringen häufig nicht den erhofften Erfolg. Umso wichtiger ist es, bei jedem Patienten nach der genauen Ätiologie der HFpEF zu suchen, um Komplikationen antizipieren und bei entsprechender Indikation eine der wenigen zielgerichteten Therapien einleiten zu können.
Die kardiale Amyloidose (CA) ist eine Ätiologie der HFpEF, die sich durch ihren fortschreitenden Charakter auszeichnet und in allen Fällen zu Komplikationen wie Erregungsleitungsstörungen, Arrhythmien und terminaler Herzinsuffizienz führt. Durch eine frühzeitige und korrekte Diagnosestellung kann der Patient einer spezialisierten Versorgung und gegebenenfalls einer spezifischen Therapie zugeführt werden, durch die sich die Krankheitsprogression verlangsamen lässt.
Kardiale Amyloidose – was ist das?
Die CA tritt im Kontext einer systemischen infiltrativen Erkrankung auf, bei der sich Proteine im Myokard ablagern und zu einer restriktiven Kardiomyopathie führen. Bei den Proteinen handelt es sich entweder um Immunglobulin-Leichtketten (im Rahmen einer monoklonalen Gammopathie von einem Plasmazell-Klon produziert; die Erkrankung wird dann als Leichtketten- bzw. AL-Amyloidose bezeichnet) oder um Transthyretin (TTR), ein von der Leber gebildetes Plasma-Transportprotein (ATTR-Amyloidose). Im Fall der Amyloidose vom Typ ATTRm (mutiertes TTR) ist das Transthyretin mutiert und defekt; im Fall der Amyloidose vom Typ ATTRwt (Wildtyp-TTR) ist es nicht mutiert. Die anderen Amyloidoseformen, insbesondere die AA-Amyloidose (Ablagerung von Amyloid A), die mit chronischen Entzündungserkrankungen einhergeht, rufen nur sehr selten Kardiomyopathien hervor. Die Amyloidfibrillen lagern sich in zahlreichen Organen ab, ohne dass dies zwangsläufig klinische Auswirkungen hat. Bei der AL-Amyloidose stehen in der Regel Schädigungen der Nieren und/oder des Herzens im Vordergrund, während es bei der ATTR-Amyloidose Schädigungen von Bändern/Sehnen, im Bereich des peripheren Nervensystems und/oder des Herzens sind. Die ATTRwt-Form ist mit Abstand die häufigste. Von ihr sind in der Regel Patienten >65 Jahre und häufiger Männer betroffen, während die anderen Formen der CA bereits in jungen Jahren auftreten können und bei beiden Geschlechtern ähnlich häufig anzutreffen sind.2
Mit den aktuellen bildgebenden Verfahren lässt sich die Krankheit zuverlässig diagnostizieren, und durch ihre systematische Anwendung konnte nachgewiesen werden, dass die Prävalenz der ATTRwt-Form deutlich höher ist als ursprünglich angenommen. Studien zeigen, dass die Prävalenz der kardialen Amyloidose bei HFpEF-Patienten bei mehr als 10% liegt.2−4 Ähnliches ist bei älteren Patienten mit Aortenklappenstenose festzustellen.5,6
Wer sollte auf eine kardiale Amyloidose untersucht werden?
Bestimmte klinische und paraklinische Merkmale wurden als «Red Flags» identifiziert, die Allgemeinmediziner oder Kardiologen zum Verdacht auf eine kardiale Amyloidose führen können, insbesondere bei Patienten >65 Jahre mit HFpEF, Aortenklappenstenose oder linksventrikulärer Hypertrophie:
-
beidseitiges Karpaltunnelsyndrom
-
verengter lumbaler Spinalkanal
-
periphere Polyneuropathie oder idiopathische Dysautonomie
-
normaler arterieller Blutdruck oder orthostatische Hypotonie nach früherer Hypertonie
-
EKG mit Mikrovoltage, fehlenden R-Zacken in präkordialen Ableitungen oder Pseudo-Q-Zacken (ohne Hinweis auf einen Infarkt) (Abb. 1)
-
Echokardiografie mit linksventrikulärer Hypertrophie (Septum >12mm), diastolischer Dysfunktion, atrialer Dilatation, dezentem Perikarderguss (Abb. 2) und verminderter longitudinaler Kontraktion mit Aussparung der apikalen Segmente (Abb. 3)

Abb. 1: EKG mit Vorhofflimmern bei einem 79-jährigen Patienten mit ATTRwt-Amyloidose. Zu beachten sind das Vorliegen eines linksanterioren Hemiblocks, von Q-Zacken in den Ableitungen II, III und aVF (Pseudoinfarkt) und die fehlende R-Progression von V1 zu V6

Abb. 2: Transthorakale Echokardiografie. Apikaler Vierkammerblick: konzentrische biventrikuläre Hypertrophie (blaue Pfeile) und biatriale Dilatation (*)
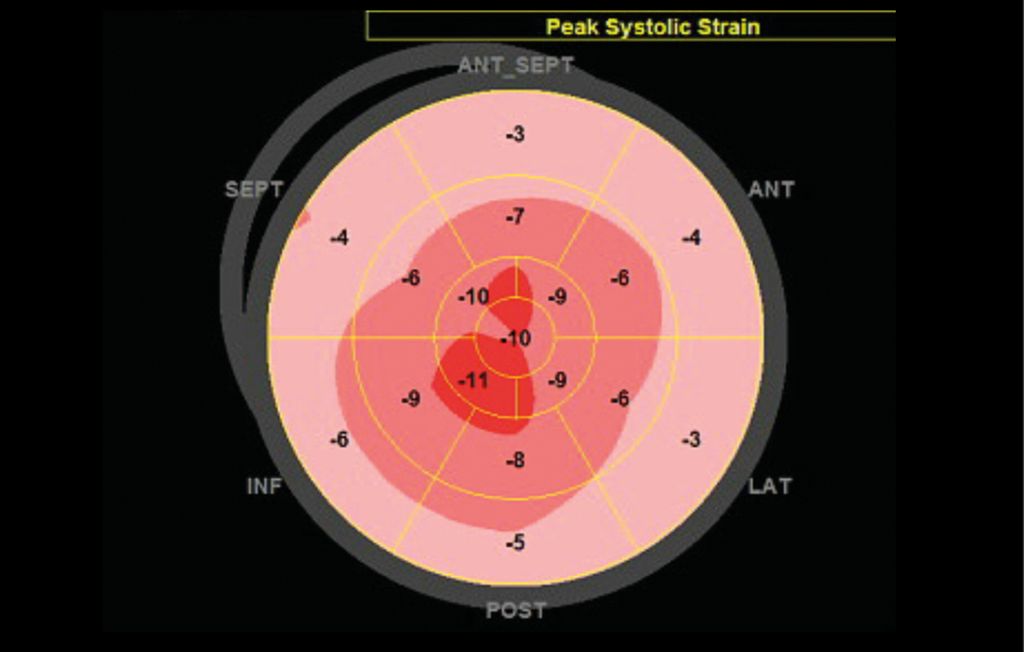
Abb. 3: Bull’s-Eye-Darstellung. Diese Darstellung zeigt die systolischen longitudinalen Strain-Werte (longitudinale Deformation) des linken Ventrikels für alle Myokardsegmente. Der Wert gibt Auskunft über die prozentuale Verkürzung der longitudinalen Fasern in der Systole. Auffällig ist ein stark reduzierter Strain in den basalen und mittleren Segmenten (Rand der Darstellung, blassrosa), während der Strain in den apikalen Segmenten weniger stark herabgesetzt ist (Mitte der Darstellung, rot); diese Verteilung ist charakteristisch für die kardiale Amyloidose
Diagnostischer Algorithmus
Das «Swiss Amyloidosis Network» (SAN), eine nationale multidisziplinäre Arbeitsgruppe, hat 2021 sehr detaillierte Empfehlungen veröffentlicht,7 die insbesondere einen standardisierten diagnostischen Ansatz vorsehen, der grob dem international etablierten Algorithmus folgt (Abb. 4).
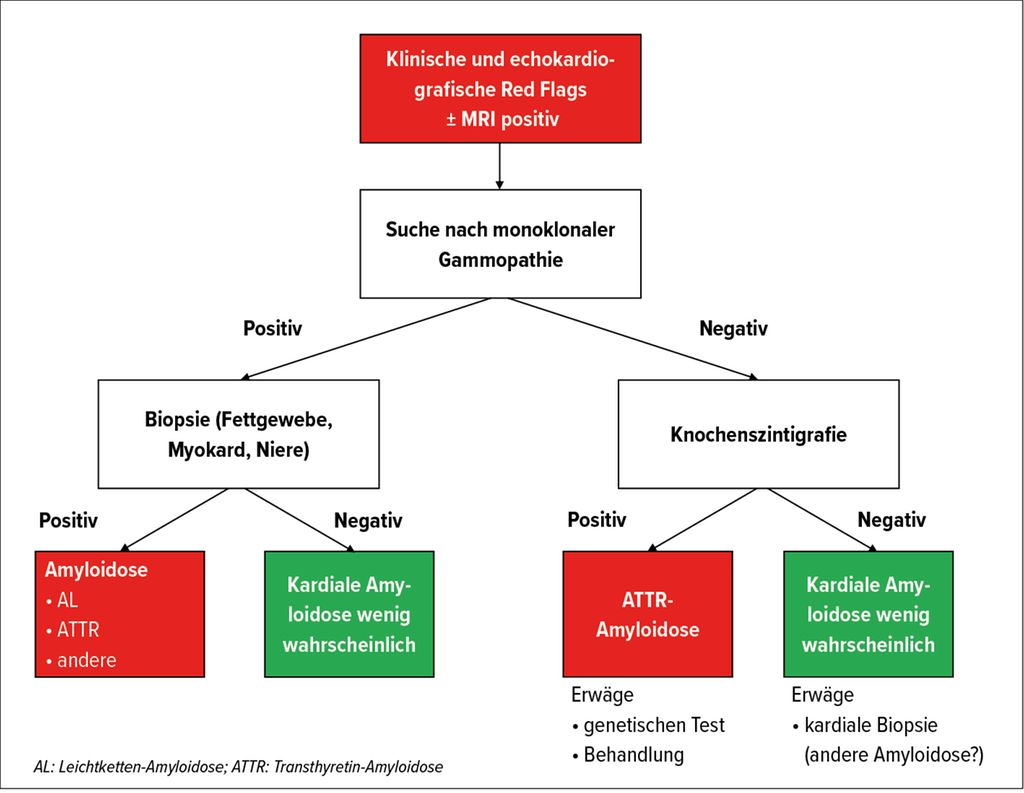
Abb. 4: Diagnostischer Algorithmus für die kardiale Amyloidose (mod. nach Hugelshofer et al.)23
Im Gegensatz zur transthorakalen Echokardiografie (TTE) und Szintigrafie/SPECT ist die Kardio-MRT für die Diagnostik nicht unverzichtbar. Insbesondere wenn Zweifel bestehen, ergänzt sie die Echokardiografie, bevor der gesamte diagnostische Algorithmus durchlaufen wird. Sie ermöglicht eine genauere Charakterisierung der Morphologie und der systolischen Ventrikelfunktion, vor allem aber den Nachweis der Infiltration des Myokards mit Amyloidfibrillen, die sich in Form einer Kontrastmittelanreicherung in Spätaufnahmen zeigt. Das Ausmass des Late-Gadolinium Enhancement (LGE) spiegelt den Schweregrad der Infiltration wider (Abb. 5).8 Noch genauer quantifizieren lässt sich diese jedoch durch Messung des myokardialen extrazellulären Volumens mithilfe einer T1-Mapping-Sequenz. Ein extrazelluläres Volumen >40% deutet dabei auf eine schwere Infiltration hin und ist sehr suggestiv für das Vorliegen einer Amyloidose.9

Abb. 5: Kardio-MRT. Mittventrikulärer Kurzachsenblick in der Spätaufnahme. In dieser Sequenz erscheint das gesunde Myokard gleichmässig schwarz. Bei diesem Patienten fällt ein zirkumferenzielles, subendokardial betontes Enhancement auf (blaue Pfeile), das sich auch auf den rechten Ventrikel erstreckt (gelbe Pfeile)
Der entscheidende Schritt in der Diagnostik ist der Ausschluss eines neoplastischen Ursprungs (AL-Typ), indem in Blut und Urin aktiv nach einer monoklonalen Gammopathie gesucht wird (Immunfixation der Proteine und Bestimmung der freien Leichtketten). Aber auch bei Hinweisen auf eine monoklonale Gammopathie muss das Vorhandensein von Amyloidablagerungen durch die Biopsie eines betroffenen Organs bestätigt werden. Da die Prävalenz der monoklonalen Gammopathie unklarer Signifikanz (MGUS) bei älteren Patienten hoch ist (5,3% bei >70-Jährigen),10 könnte bei diesen ohne histologische Untersuchung das Bestehen einer ATTRwt-Amyloidose in Kombination mit einer MGUS sonst leicht mit einer AL-Amyloidose verwechselt werden. In diesem Fall bestünde das Risiko einer Fehlbehandlung der Patienten.
Die Ganzkörper-Szintigrafie mittels SPECT (Single Photon Emission Computer Tomography) unter Verwendung eines Tracers (in der Schweiz häufig 99mTc-DPD) ist fester Bestandteil der Diagnostik der ATTR-Amyloidose. Bei Bestehen einer Amyloidose lagert sich der Tracer in die Mikrokalzifikationen in den Amyloidfibrillen ein und reichert sich im Myokard an (Abb. 6). Dieses Verfahren bietet somit eine hohe Sensibilität für den Nachweis einer CA vom ATTR-Typ,11 auch wenn seine Spezifität nicht optimal ist, da die Szintigrafie in etwa einem Drittel der AL-Amyloidose-Fälle schwach positiv ausfällt.12 Zu einem falsch positiven Ergebnis kann die Untersuchung führen, wenn kürzlich ein ausgedehnter Infarkt stattgefunden hat, wenn eine einfache Szintigrafie (planare Aufnahmen ohne SPECT) durchgeführt wird und die Anreicherung des Tracers im «blood pool» mit einem «uptake» im Myokard verwechselt wird oder wenn der seltene Fall einer Chloroquin-bedingten toxischen Kardiomyopathie vorliegt. Dennoch kann nach sorgfältigem Ausschluss einer monoklonalen Gammopathie eine positive Skelettszintigrafie im Bereich des Myokards als Nachweis einer ATTR-Amyloidose betrachtet werden. Umgekehrt ist eine CA im Falle einer negativen Szintigrafie und des Fehlens einer monoklonalen Gammopathie nahezu ausgeschlossen. In solchen Fällen bleibt die Myokardbiopsie Situationen vorbehalten, in denen klinisch nach wie vor ein starker Verdacht auf eine Amyloidose besteht.
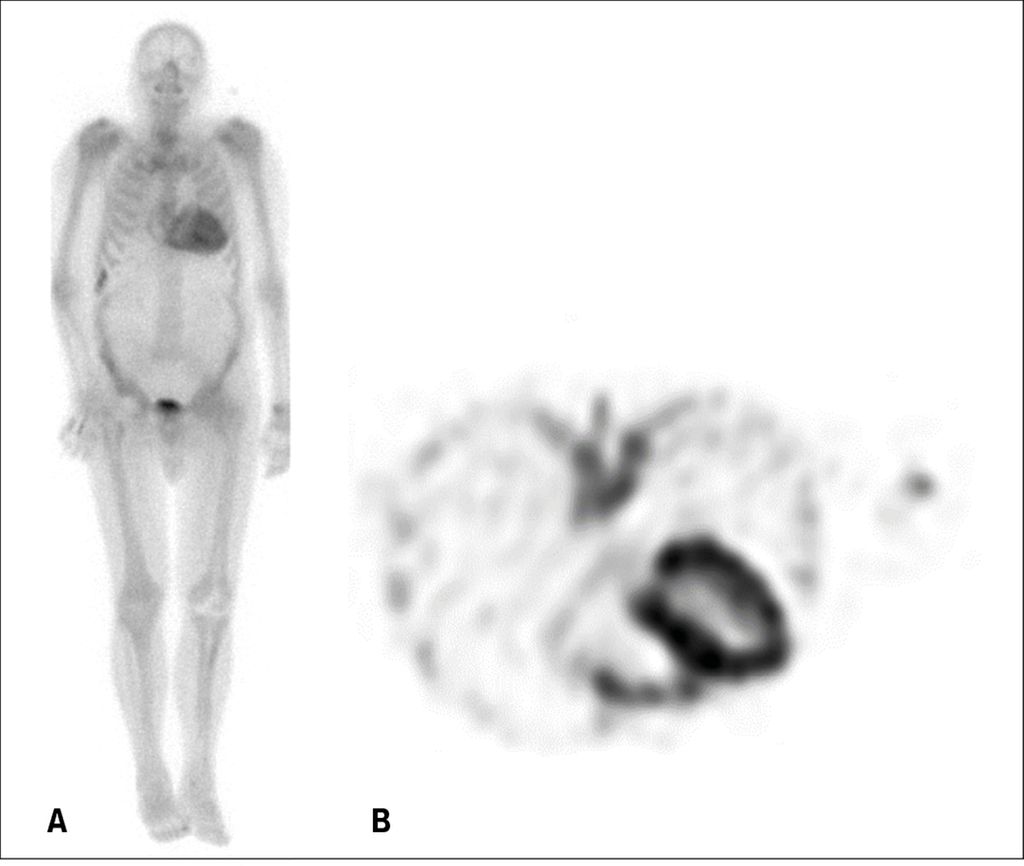
Abb. 6: Skelettszintigrafie/SPECT mit 99mTc-DPD (Beclometasondipropionat) bei einem Patienten mit kardialer Amyloidose vom ATTR-Typ. A) Planare Aufnahme; B) SPECT-Aufnahme
Auch wenn eine familiäre Amyloidose (ATTRm) nur selten bei >65-Jährigen festgestellt wird, wird bei allen Patienten mit ATTR-CA unabhängig von ihrem Alter nach Stellung der Diagnose ein Gentest empfohlen. Dieser ist gerechtfertigt, da das Vorliegen einer Mutation direkte Folgen für Verwandte ersten Grades hat.
Prognose
Im Allgemeinen hängt die Prognose jeder systemischen Amyloidose stark davon ab, ob eine kardiale Infiltration vorliegt oder nicht. Besteht eine solche Infiltration, hat die Form der Amyloidose Einfluss auf die Prognose. Das mediane Überleben ohne Behandlung liegt bei einer CA vom AL-Typ bei 5,4 Monaten, bei einer CA vom TTR-Typ dagegen bei 60 Monaten.8,13 Auch wenn die individuelle Einschätzung der Prognose im Wesentlichen auf der Bestimmung kardialer Biomarker (TroponinT und NT-proBNP) beruht, haben sich der Schweregrad der longitudinalen Dysfunktion des linken Ventrikels in der Echokardiografie14 und das Ausmass des LGE in der Kardio-MRT8 als unabhängige Faktoren mit prognostischem Wert erwiesen.
Therapie
Es lassen sich zwei Gruppen von Therapien unterscheiden: einerseits die symptomatischen Therapien der Herzinsuffizienz und andererseits Therapien, die das Ziel verfolgen, die Krankheitsprogression zu verlangsamen und die Prognose zu verbessern («disease modifying treatments»).
Symptomatische Therapien
Die CA ist eine Form einer restriktiven Kardiomyopathie, die durch eine kleine linksventrikuläre Kavität mit geringer Compliance gekennzeichnet ist. Aus diesem Grund ist eine erhaltene atriale Kontraktion wichtig, um die Ventrikelfüllung trotz der diastolischen Dysfunktion zu optimieren. Um die Symptome, allen voran die Dyspnoe, zu lindern, gilt es deshalb zunächst, die Volämie zu optimieren und einen Sinusrhythmus aufrechtzuerhalten. Da ein steifes Myokard nicht mehr in der Lage ist, sich an Schwankungen der Vorlast anzupassen, kann durch eine regelmässig überprüfte diuretische Therapie eine ausreichende Ventrikelfüllung aufrechterhalten werden. In fortgeschrittenen Fällen ist die therapeutische Breite jedoch gross und das Finden des richtigen Gleichgewichts schwierig: Häufig schwanken die Patienten zwischen Volumenüberlastung und Niereninsuffizienz. Die Infiltration der Vorhöfe und der erhöhte Druck führen systematisch zu einer atrialen Dilatation und häufig zu Arrhythmien (Vorhofflimmern, -flattern). Die Strategie zur Rhythmuskontrolle muss so lange wie möglich weiterverfolgt werden, da die Patienten im Sinusrhythmus weniger Symptome zeigen. Tritt das Vorhofflimmern trotzdem erneut auf, ist eine vorsichtige Kontrolle der Ventrikelfrequenz indiziert, wobei Betablocker die Erstlinientherapie darstellen. Sowohl Digoxin als auch Kalziumantagonisten verbinden sich mit den Amyloidfibrillen und reichern sich in den infiltrierten Geweben an, wodurch sich das Risiko toxischer Arzneimittelwirkungen erhöht.15,16 Sobald eine atriale Arrhythmie diagnostiziert wird, ist es sehr wichtig, eine orale Antikoagulation zu verordnen, da unabhängig vom CHA2DS2-VASc-Score ein erhöhtes Risiko für einen kardioembolischen Schlaganfall besteht.
Die Infiltration der Leitungsbahnen bei einer Amyloidose kann auch einen mehr oder weniger schweren atrioventrikulären Block (AVB) zur Folge haben. Für die Implantation eines Herzschrittmachers gelten die gleichen Empfehlungen wie für den AVB degenerativen Ursprungs. Weniger gut etabliert ist die Behandlung jedoch, wenn es um die Prävention des plötzlichen Herztods geht. Auch wenn Herzrhythmusstörungen in dieser Population eine häufige Todesursache zu sein scheinen, konnte nicht gezeigt werden, dass dieses Risiko durch einen primärpräventiv implantierten Defibrillator gesenkt werden kann.17
Krankheitsmodifizierende Therapien
Die Behandlung der AL-Amyloidose besteht in der Therapie des multiplen Myeloms und erfolgt durch Spezialisten für Hämatologie/Onkologie. Auf diese komplexe Therapie (Chemotherapie ± Knochenmarktransplantation) wird in diesem Artikel nicht näher eingegangen.
Die einzige Behandlungsoption für die ATTR-CA war lange Zeit die Lebertransplantation, die jungen Patienten mit ATTRm-CA vorbehalten war. In den letzten Jahren haben sich jedoch neue Substanzen als wirksam erwiesen. Der erste in der Schweiz zur Behandlung der ATTR-CA erhältliche Wirkstoff ist Tafamidis (Vyndaqel®), welches das Protein TTR stabilisiert, damit es seine Tetramerstruktur behält. Eine randomisierte Studie mit >400 Patienten zeigte nach 30 Monaten der Behandlung einen signifikanten Nutzen in Bezug auf die Mortalität (absolute Reduktion um 13%), die Hospitalisierungen (absolute Reduktion um 32%) sowie die Lebensqualität.18 Der Nutzen scheint bei weniger weit fortgeschritter Erkrankung (NYHA-Klasse I–II) höher zu sein. Auf Grundlage dieser Studie wurde Tafamidis im Dezember 2021 in die Spezialitätenliste des BAG aufgenommen, mit der Limitation, dass es in der Schweiz nur von kardiologischen Weiterbildungsstätten der Kategorie A verordnet werden darf (Abb. 7). Die Kosten der Behandlung werden nur bei Patienten übernommen, die die folgenden Kriterien erfüllen: Diagnose einer ATTR-CA entsprechend dem Algorithmus (Abb.4), ≥1 akute Episode einer Herzinsuffizienz, die die Anwendung von Diuretika erforderte, NYHA-Klasse I–II, NT-proBNP-Konzentration >600pg/ml, Distanz im 6-Minuten-Gehtest >100m, Lebenserwartung von mindestens zwei Jahren.19
Abb. 7: Kardiologische Weiterbildungsstätten der Kategorie A in der Schweiz
Dabei ist zu beachten, dass das Medikament mit einem Preis von >10000Fr./Monat im Moment noch sehr teuer ist. Deshalb ist es äusserst wichtig, die Indikation für die Behandlung zwischen Patient, Hausarzt und Spezialist zu besprechen, bevor ein Kostengutsprachegesuch gestellt wird bzw. in manchen Fällen sogar bevor der diagnostische Algorithmus durchlaufen wird.
In den vergangenen Jahren wurde noch eine weitere Gruppe von Medikamenten entwickelt, die als «TTR gene silencers» bezeichnet werden und deren Wirkprinzip in der Unterbrechung der TTR-Synthese auf molekularer Ebene besteht. Aktuell existieren zwei Wirkstoffe, Patisiran20 und Inotersen,21 die in der Schweiz erhältlich und zur Behandlung von Polyneuropathien bei familiärer Amyloidose (TTRm) indiziert sind. In einer vordefinierten Subgruppe von Patienten mit Herzerkrankung konnte gezeigt werden, dass Patisiran zu einer Verbesserung der systolischen Funktion des linken Ventrikels und einer Reduktion der NT-proBNP-Werte führt.22 Allerdings werden die Kosten der Behandlung bei CA bisher nicht übernommen. Verschiedene Phase-III-Studien laufen aktuell, und es ist damit zu rechnen, dass das therapeutische Arsenal zur Behandlung der CA in den kommenden Jahren durch weitere Wirkstoffe erweitert werden wird.
Literatur:
1 McDonagh T et al.: 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2021; 42: 3599-726 2 Gonzalez-Lopez E et al.: Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36: 2585-94 3 Hahn VS et al.: Endomyocardial biopsy characterization of heart failure with preserved ejection fraction and prevalence of cardiac amyloidosis. JACC Heart Fail 2020; 8: 712-24 4 Bennani-Smires Y et al.: Pilot study for left ventricular imaging phenotype of patients over 65 years old with heart failure and preserved ejection fraction: the high prevalence of amyloid cardiomyopathy. Int J Cardiovasc Imaging 2016; 32: 1403-13 5 Castano A et al.: Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 2017; 38: 2879-87 6 Longhi S et al.: Coexistence of degenerative aortic stenosis and wild-type transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging 2016; 9: 325-7 7 Condoluci A et al.: Management of transthyretin amyloidosis. Swiss Med Wkly 2021; 151: w30053 8 Fontana M et al.: Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2015; 132: 1570-9 9 Banypersad SM et al.: Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: an equilibrium contrast cardiovascular magnetic resonance study. Circ Cardiovasc Imaging 2013; 6: 34-9 10 Dispenzieri A et al.: Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet 2010; 375: 1721-8 11 Gillmore JD et al.: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016; 133: 2404-12 12 Rapezzi C et al.: Systemic cardiac amyloidosis disease profile and clinical courses of the three main types. Circulation 2009; 120: 1203-12 13 Kumar S et al.: Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012; 30: 989-95 14 Buss SJ et al.: Longitudinal left ventricular function for prediction of survival in systemic light-chain amyloidosis: incremental value compared with clinical and biochemical markers. J Am Coll Cardiol 2012; 60: 1067-76 15 Muchtar E et al.: Digoxine use in systemic light chain amyloidosis – contra-indicated or cautious use? Amyloid 2018; 25: 86-92 16 Gertz M, Rodney H et al.: Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol 1985; 55: 1645 17 Lyn D et al.: Implantable cardioverter defibrillators in patients with cardiac amyloidosis. J Cardiovasc Electrophysiol 2013; 24: 793-8 18 Maurer MS et al.: Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379: 1007-16 19 Liste des spécialités, Office fédéral de la santé publique OFSP, octobre 2022 20 Adams D et al.: Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 11-21 21 Benson MD et al.: Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 22-31 22 Solomon S et al.: Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation 2019; 139: 431-43 23 Hugelshofer S et al.: Cardiac amyloidosis: a rare disease? Rev Med Suisse 2019; 15: 1054-9
Das könnte Sie auch interessieren:

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

Inclisiran bei Patienten mit Statinintoleranz wirksam und sicher
Eine Analyse statinintoleranter Patienten aus dem Phase III Studienprogramm ORION zeigt, dass Inclisiran die LDL-Cholesterinspiegel kardiovaskulärer Hochrisikopatienten, die kein Statin ...