
Kardiomyopathien: phänotypbasierte Einteilung, Ursachensuche, Management
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die Leitlinie zum Management von Kardiomyopathien wurde 2023 auf dem Kongress der Europäischen Gesellschaft für Kardiologie (ESC) präsentiert. Sie beinhaltet neue Empfehlungen zu Diagnostik, Risikostratifizierung und Behandlung von Kardiomyopathien, die sich in den klinischen Alltag integrieren lassen.
Keypoints
-
Bei jeder CMP-Erstdiagnose soll nach Festlegung des kardialen Phänotyps die Suche nach einem bestimmten Auslöser eingeleitet werden.
-
Die kardiale Magnetresonanztomografie wird bei jeder CMP-Erstdiagnose empfohlen und soll im Intervall wiederholt werden.
-
Die genetische Testung bzw. das Familienscreening fließen in das CMP-Management ein (Screening, Diagnose, Risikostratifizierung, Follow-up).
Die Europäische Gesellschaft für Kardiologie veröffentlichte 2023 die erste internationale Leitlinie zum Management von Kardiomyopathien (CMP). Abgesehen von Empfehlungen zur hypertrophen CMP, die im Jahr 2014 publiziert und nun aktualisiert wurden, handelt es sich zum größten Teil um neue Empfehlungen. Mangels randomisierter, kontrollierter Studien stützen sich die CMP-Guidelines überwiegend auf Expertenmeinungen (65% Evidenzgrad C). In den ESC-Leitlinien hervorgehoben werden insbesondere
-
die Suche nach einem bestimmten Auslöser der CMP,
-
die kardiale Magnetresonanztomografie (CMR), die bei allen CMP-Patient:innen bei Erstdiagnose empfohlen wird, und
-
die genetische Testung bzw. das Familienscreening.
Was sind Kardiomyopathien?
CMP beschreiben Herzmuskelerkrankungen, die durch strukturelle oder funktionelle Abnormalitäten des Herzens gekennzeichnet sind und nicht ausschließlich auf eine koronare Herzkrankheit, eine arterielle Hypertonie, eine Herzklappenerkrankung oder einen angeborenen Herzfehler zurückzuführen sind.
Festlegung des kardialen Phänotyps
Der Verdacht auf CMP kann aufgrund von Symptomen/klinischen Zeichen, abnormen Zufallsbefunden oder einer positiven Familienanamnese erhoben werden. Der diagnostische Pfad beginnt mit der Festlegung des kardialen Phänotyps, der sich aus morphologischen und funktionellen Merkmalen ergibt (Abb. 1):
-
hypertrophe CMP (HCM)
-
dilatative CMP (DCM)
-
nichtdilatative linksventrikuläre CMP (NDLVC)
-
arrhythmogene rechtsventrikuläre CMP (ARVC)
-
restriktive CMP (RCM)
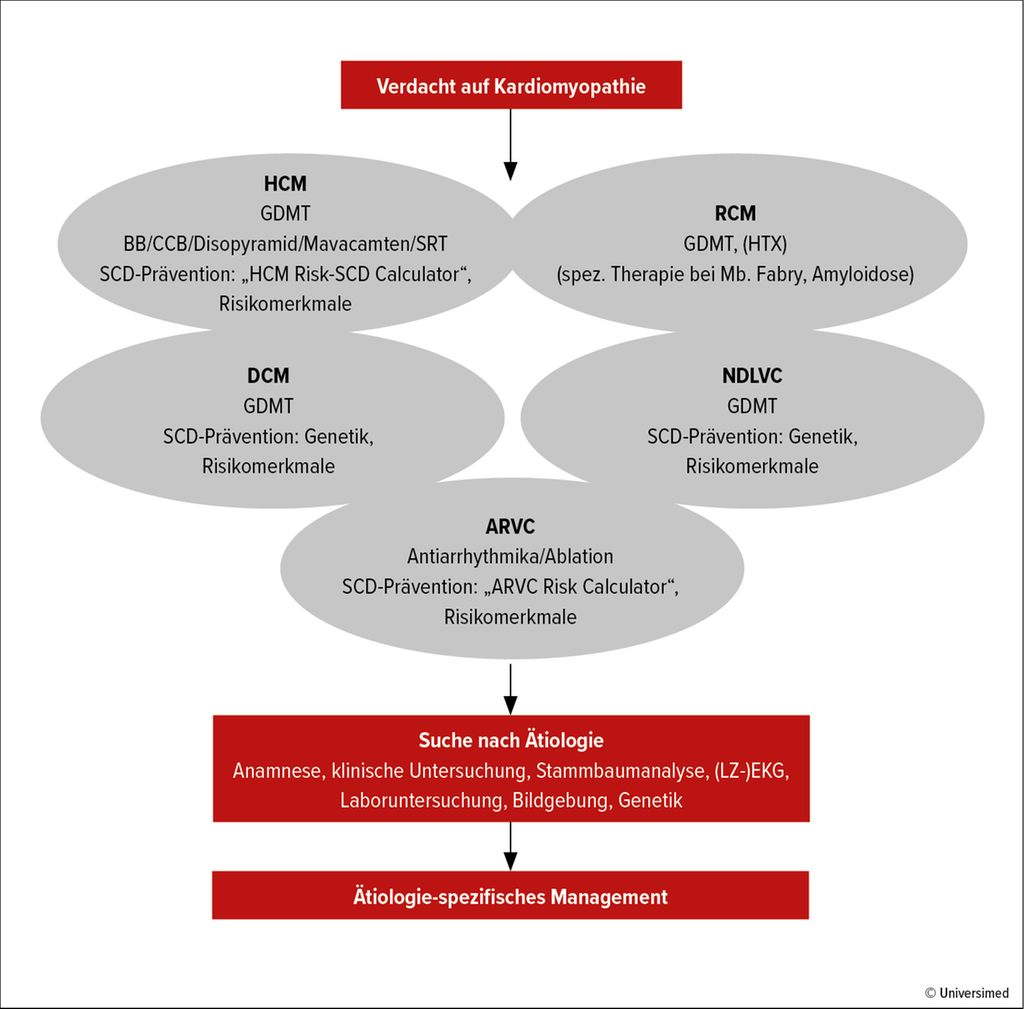
Abb.1: Vorgehen bei Verdacht auf Kardiomyopathie (mod. nach Arbelo E et al., Eur Heart J 2023)
Im Anschluss soll die Suche nach einem bestimmten Auslöser der CMP eingeleitet werden: Anamnese, klinische Untersuchung, Stammbaumanalyse über 3–4 Generationen, EKG, Langzeit-EKG, Laboruntersuchung, Bildgebung. Die Einbindung mehrerer Fachdisziplinen in die CMP-Abklärung ist von entscheidender Bedeutung, um komplexen syndromalen Erkrankungen gerecht zu werden.
Bildgebung
Bildgebende Verfahren spielen eine entscheidende Rolle, wobei die Durchführung einer CMR bei jeder CMP-Erstdiagnose empfohlen wird. Weitere Untersuchungen wie Skelettszintigrafie (Amyloidose), FDG-PET (Sarkoidose) oder CT (Koronaranomalien) können ebenso erforderlich sein. In begründeten Fällen kann auch eine Endomyokardbiopsie durchgeführt werden.
Genetik
Nach der Initialdiagnostik wird die genetische Testung durchgeführt. Diese wird in der Leitlinie umfangreich abgehandelt und es gibt Empfehlungen, wer wann getestet werden soll. Das Ergebnis der Gentestung soll auch in die Prognoseeinschätzung einfließen, woraus sich entsprechende therapeutische Konsequenzen ableiten (u.a. ICD-Implantation). Indexpatient:in und Familienangehörige sollen begleitend humangenetisch beraten werden.
CMP-Phänotypen und Management
Wenngleich eine CMP-Erstdiagnose die Suche nach einer bestimmten Ursache auslösen soll, spricht die aktuelle ESC-Leitlinie keine detaillierten Empfehlungen zum Management spezifischer CMP-Ursachen aus. Hingegen wird auf das allgemeine Management von CMP eingegangen.
Hypertrophe Kardiomyopathie
Die Empfehlungen zur HCM stammen bereits aus dem Jahr 2014. Diese wurden nun aktualisiert und in die aktuelle Leitlinie integriert. Eine neue Empfehlung betrifft die medikamentöse Therapie der linksventrikulären Ausflusstraktobstruktion (LVOTO). Obwohl Betablocker (BB) und Kalziumkanalblocker (CCB) nach wie vor die Therapien der ersten Wahl sind, wird erstmals der kardiale Myosin-Inhibitor Mavacamten als spezifische medikamentöse Behandlungsoption als Zweitlinientherapie aufgenommen.
Eine septale Reduktionstherapie (SRT) in Form einer Alkoholseptumablation oder Myektomie kann bei folgenden Konstellationen erfolgen:
-
Symptome (NYHA III/IV) trotz maximaler medikamentöser Therapie,
-
NYHA II/LVOTO ≥50mmHg in Verbindung mit zumindest mittelgradiger Mitralklappeninsuffizienz, Vorhofflimmern oder linksatrialer Dilatation.
Für die Primärprävention des plötzlichen Herztodes (SCD) wird weiterhin die Verwendung des „HCM Risk-SCD Calculators“ empfohlen. Daraus errechnet sich das 5-Jahres-Risiko für einen plötzlichen Herztod (SCD), woraus sich Empfehlungen zur primärprophylaktischen ICD-Implantation ableiten.
Fallbeispiel einer 54-jährigen Patientin
Eine 54-jährige Patientin präsentiert sich mit zunehmender Belastungsdyspnoe (NYHA III). Im EKG zeigt sich der Sokolow-Lyon-Index positiv als Hinweis für eine Linksherzhypertrophie, zusätzlich finden sich HCM-charakteristische Repolarisationsstörungen bei tief-negativen T-Wellen. Echokardiografisch bestätigt sich der hypertrophe Phänotyp und eine LVOTO in Ruhe (>50mmHg) und unter Valsalva (>100mmHg) kann erfasst werden. In der CMR ergibt sich kein Hinweis auf eine myokardiale Speichererkrankung. Laborchemisch zeigen sich erhöhte kardiale Biomarker; die Leichtkettendiagnostik verläuft negativ. Nach einer umfassenden Diagnostik wird bei ihr eine hypertroph-obstruktive Kardiomyopathie (HOCM) diagnostiziert. Die genetische Testung ergibt eine MYH7-Genmutation (negative Familienanamnese). Auf Basis der positiven Gentestung wird ein Familienscreening eingeleitet. Im LZ-EKG können keine Arrhythmien nachgewiesen werden. Das 5-Jahres-SCD-Risiko liegt bei <4% (keine primärprophylaktische ICD-Implantation, Reevaluierung in 1–2 Jahren).
Die BB-Prämedikation wird dosisgesteigert und mangels Symptomkontrolle auf eine CCB-Therapie umgestellt. Da sich auch hierunter keine Besserung einstellt, wird Mavacamten als Zweitlinientherapie versucht und die Patientin engmaschig klinisch und echokardiografisch verlaufskontrolliert. Zeitnah kann eine Beschwerdebesserung (von NYHA III auf II) auf Basis einer LVOTO-Reduktion (von Ruhe-LVOTO >50mmHg auf 27mmHg) erreicht werden. Im Falle fortbestehender Symptome hätte eine SRT erwogen werden können.
Dilatative Kardiomyopathie
Die DCM kann mit einem erhöhten arrhythmogenen Risiko bei Vorliegen einer Hochrisiko-Genkonstellation einhergehen: u.a. LMNA, FLNC-truncating-Varianten, TMEM43, PLN, DSP und RBM20. Neben dem Genstatus werden zusätzliche Risikofaktoren berücksichtigt, um über eine primärprophylaktische ICD-Implantation zu entscheiden: u.a. CMR: „extensive LGE (late gadolinium enhancement)“, LVEF <45%, nichtanhaltende ventrikuläre Tachykardien (nsVT), häufige ventrikuläre Extrasystolen (VES), Synkopen.
Fallbeispiel eines 43-jährigen Patienten
Ein 43-jähriger Patient erleidet einen Herz-Kreislauf-Stillstand. Nach kurzer Laienreanimation und unmittelbarer Schockabgabe bei Kammerflimmern kann ein Wiedereinsetzen des Spontankreislaufs nach dem Herz-Kreislauf-Stillstand (ROSC) erreicht werden. Der Patient galt bislang als gesund und gut leistungsfähig und es gab eine negative Familienanamnese bzgl. SCD. Bei Eintreffen im Schockraum erfolgt nach Stabilisierung des Patienten und Ausschluss reversibler Ursachen eine TTE, wobei eine hochgradig reduzierte systolische LV-Funktion (LVEF ca. 25%) bei einem dilatativen Phänotyp erfasst werden kann. Die klinische Untersuchung verläuft unauffällig und das EKG weist bis auf einzelne VES keine Besonderheiten auf. Eine im Anschluss durchgeführte Schädel/Körperstamm-CT-Untersuchung ergibt abgesehen von einigen Reanimationsverletzungen keine Befunderweiterung. In der Koronarangiografie kann eine stenosierende KHK ausgeschlossen werden.
Auf Basis der HFrEF wird nach erreichter hämodynamischer Stabilisierung rasch eine HI-Quadrupeltherapie initiiert (RAAS-Blockade/BB/MRA/SGLT2-Inhibitor). Die CMR zeigt neben der eingeschränkten LV-Funktion ein ausgedehntes LGE (septal mid-wall), vereinbar mit einer Laminopathie. Eine genetische Testung sichert die Diagnose einer LMNA-assoziierten dilatativen Kardiomyopathie. Es erfolgen eine sekundärprophylaktische ICD-Implantation, ein Familienscreening und mangels spezifischer Therapiemöglichkeiten eine Fortführung der HI-Quadrupeltherapie. Hierunter gelingt eine Besserung der systolischen LV-Funktion auf 45%. Von übermäßiger körperlicher Anstrengung wird dem Patienten abgeraten.
Nichtdilatative linksventrikuläre Kardiomyopathie
Die Einführung einer neuen Phänotyp-Kategorie erfolgt aufgrund des Vorhandenseins zahlreicher Zwischenformen, die historisch noch nicht alle Kriterien einer festgelegten Phänotyp-Klasse erfüllen. Die Gruppe der NDLVC umfasst:
-
Patient:innen, bei denen in der Bildgebung eine nichtischämische Narbe festgestellt wird, mit oder ohne funktionelle Einschränkung, und
-
Patient:innen mit einer systolischen Dysfunktion ohne Narbenbildung.
Die Empfehlungen betreffend die ICD-Implantation bei NDLVC entsprechen jenen für die DCM.
Arrhythmogene rechtsventrikuläre Kardiomyopathie
Praxistipp
Jede Kardiomyopathie soll die Suche nach einem Auslöser anstoßen.Die ARVC ist durch einen Kardiomyozytenverlust mit konsekutivem Ersatz durch Fett- und Bindegewebszellen gekennzeichnet. Aufgrund des erhöhten Arrhythmierisikos sind Antiarrhythmika (v.a. BB) und/oder die Katheterablation wertvolle Therapieoptionen. Von übermäßiger körperlicher Anstrengung sollte abgesehen werden. Das Ergebnis des „ARVC Risk Calculator“ und andere Risikofaktoren (z.B. Synkopen, nsVT, RVEF <40%, LVEF <45%, smVT PES) sollen in die Entscheidung für/gegen primärprophylaktische ICD-Implantation einfließen.
Restriktive Kardiomyopathie
Die RCM weist die ungünstigste Prognose auf. Es ist von entscheidender Bedeutung, zwischen einer „echten RCM“ und anderen Erkrankungen zu unterscheiden, die eine restriktive Physiologie aufweisen, aber einen anderen Phänotyp haben und daher nicht als „echte RCM“ angesehen werden sollten, z.B. Mb. Fabry, Amyloidose.
Literatur:
bei der Verfasserin
Das könnte Sie auch interessieren:

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

Inclisiran bei Patienten mit Statinintoleranz wirksam und sicher
Eine Analyse statinintoleranter Patienten aus dem Phase III Studienprogramm ORION zeigt, dass Inclisiran die LDL-Cholesterinspiegel kardiovaskulärer Hochrisikopatienten, die kein Statin ...