
Focus sur la cardiomyopathie hypertrophique – la comprendre et la traiter
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
La cardiomyopathie hypertrophique (CMH) est la cardiomyopathie héréditaire la plus fréquente, mais passe souvent inaperçue. Avec une prévalence globale de 1:500 et un risque accru d’événements cardiovasculaires, le diagnostic et le traitement de cette maladie restent un défi.
Keypoints
-
Le phénotype de la CMH peut présenter les étiologies les plus diverses. La cause la plus fréquente est une mutation dans les gènes codant pour des protéines du sarcomère cardiaque.
-
L’échocardiographie joue un rôle clé dans la description du phénotype.
-
L’intégration d’autres imageries/examens et/ou de l’analyse génétique est souvent essentielle pour poser le diagnostic ainsi que le diagnostic différentiel.
-
Le mavacamten est une «nouvelle» option thérapeutique pour la CMH avec obstruction de la chambre de chasse du ventricule gauche.
Définition
Une CMH existe en cas d’augmentation de l’épaisseur ≥15mm dans un segment myocardique, si cette augmentation ne peut pas être expliquée uniquement par des conditions de remplissage anormales telles qu’une sténose de la valve aortique ou une hypertension. Chez les parents, en cas de formes génétiques ou de modifications typiques de l’ECG, une augmentation moindre de l’épaisseur de la paroi (13–14mm) peut toutefois déjà être présente.1 Sur le plan clinique, des troubles tels qu’une dyspnée, une angine de poitrine, des vertiges ou une syncope sont décrits en plus des arythmies.
Prévalence
La prévalence globale de la CMH est de 0,2%, ce qui correspond à environ 20 millions de personnes dans le monde. Une grande partie, estimée à 85–90%, ne sont toutefois pas diagnostiquées. Environ 60% des personnes concernées développent des symptômes.2–4 Il a été démontré que les patient·es atteint·es de CMH présentent un risque accru d’événements cardiovasculaires et que la mortalité globale est environ 4 fois plus élevée que dans la population générale. Environ 62% souffrent de fibrillation atriale et le risque de «sudden cardiac death» (SCD) est de 1 à 2%.5–8
Étiologie et pathogenèse
Les phénotypes de la CMH reposent sur différents mécanismes pathologiques et étiologies. Jusqu’à 60% des patient·es atteint·es de CMH présentent une variante héréditaire autosomique dominante causée par des mutations dans les gènes codant pour des protéines du sarcomère cardiaque. Il en résulte une formation accrue de ponts actine-myosine avec hypercontractilité, diminution de la relaxation, augmentation de la rigidité et fibrose. La majorité des mutations décrites ont lieu dans les chaînes lourdes bêta de la myosine (MYH7), de la protéine C de liaison à la myosine (MYBPC3), de la troponine I (TNNI3) et de la troponine T (TNNT2). Chez environ 10% des personnes concernées, le phénotype de la CMH a des causes très diverses, les plus fréquentes étant les maladies de stockage, telles que l’amylose, les maladies de stockage lysosomales (maladie de Fabry, maladie de Danon) ou encore les maladies de stockage du glycogène. Chez environ 30% des patient·es, la cause reste inexpliquée.9
Obstruction de la chambre de chasse du ventricule gauche (LVOTO)
L’identification d’une LVOTO est importante pour la prise en charge des symptômes et l’évaluation du risque de SCD. Environ 37% des patient·es présentent une obstruction au repos, tandis que 70% ne développent une obstruction qu’après provocation.10 Le rétrécissement de la chambre de chasse est dû à l’hypertrophie et à des modifications anatomiques de la valve mitrale. Un signal de flux «dagger-shaped» au doppler continu ainsi qu’un SAM («systolic anterior motion») du feuillet antérieur de la valve mitrale sont typiques. Si le gradient est >30mmHg, une obstruction est présente.
Diagnostic
Les cardiomyopathies sont classées en cinq phénotypes différents, selon les «2023 ESC Guidelines for the management of cardiomyopathies»: cardiomyopathie hypertrophique (CMH), cardiomyopathie dilatée (CMD), cardiomyopathie ventriculaire gauche non dilatée (CVGND), cardiomyopathie arythmogène du ventricule droit (CAVD) et cardiomyopathie restrictive (CMR; Fig. 1).1
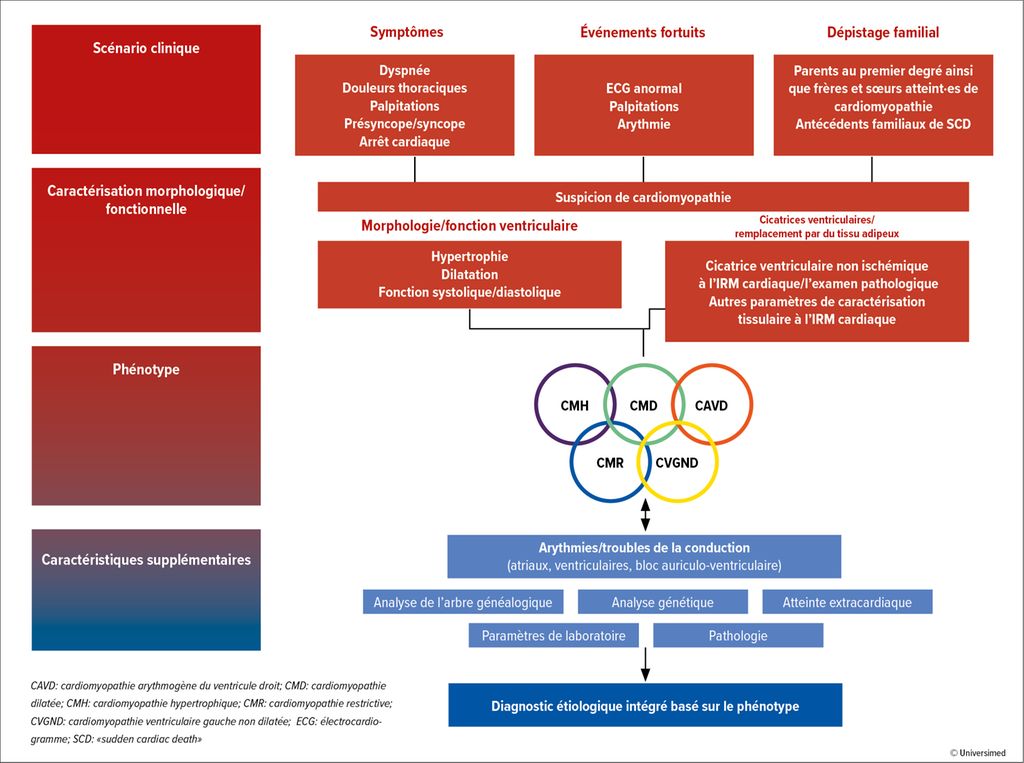
Fig.1: Approche diagnostique (modifiée selon Arbelo E et al. 2023)1
En cas d’apparition de symptômes tels que dyspnée, douleurs thoraciques, palpitations, syncopes, ainsi qu’en présence de modifications pathologiques de l’ECG ou d’anomalies auscultatoires, une imagerie telle qu’une échocardiographie et/ou une imagerie par résonance magnétique (IRM) est recommandée. En outre, des examens génétiques et des paramètres de laboratoire sont recommandés, et l’anamnèse familiale/l’arbre généalogique est recueilli.
La détermination du phénotype (CMH, CMD, CVGND, CAVD, CMR) ne constitue pas en soi un diagnostic, mais une description de la morphologie à partir de l’imagerie cardiaque. L’étiologie ou le mécanisme pathologique doit être déterminé par des examens complémentaires pour poser le diagnostic. C’est pourquoi l’approche systématique ne doit pas s’arrêter à l’imagerie. Une grande importance est accordée à l’analyse génétique, qui doit être réalisée chez l’ensemble des patient·es atteint·es de cardiomyopathie qui peuvent en tirer profit en termes de diagnostic, de pronostic et de traitement (recommandation de niveau 1B).1
Importance de l’échocardiographie
L’échocardiographie est le premier et principal outil diagnostique utilisé. Il existe des caractéristiques d’imagerie spécifiques qui peuvent indiquer une étiologie particulière de la CMH, mais qui ne permettent pas à elles seules de poser un diagnostic (Tab. 1). Dans l’échocardiographie transthoracique, outre les fonctions systolique et diastolique, la description des valves, les dimensions des cavités cardiaques et la pression artérielle pulmonaire systolique (PAPs), une attention particulière doit être accordée à la mesure exacte de l’épaisseur maximale de la paroi effectuée en fin de diastole. Il est recommandé de réaliser des mesures à partir de différentes vues de l’axe cardiaque. La distribution asymétrique de l’hypertrophie est typique de la CMH. La mesure de la masse ventriculaire gauche peut être normale jusqu’à 20% en raison de la distribution asymétrique isolée de l’hypertrophie.11
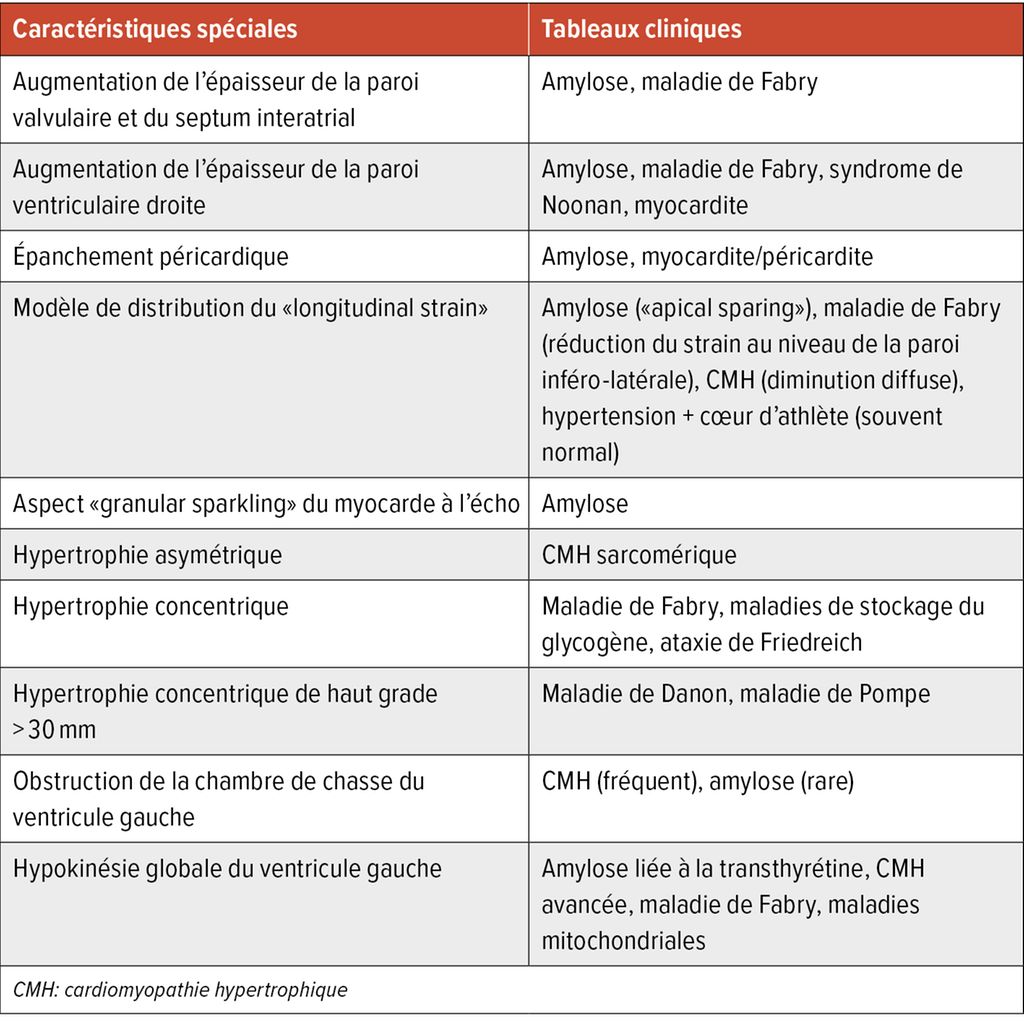
Tab.1: Caractéristiques échocardiographiques indiquant une étiologie particulière de la CMH (modifié selon Arbelo E et al. 2023)1
L’identification d’une obstruction de la chambre de chasse du ventricule gauche est importante pour la prise en charge des symptômes et l’évaluation du risque de SCD. Une analyse 2D pour identifier un SAM et une mesure par doppler doivent être effectuées au repos et après manœuvre de Valsalva ou en situation de stress. En cas de conditions échographiques restreintes et/ou de suspicion de CMH apicale, l’utilisation d’un produit de contraste, à gauche, s’avère utile. Dans un travail de Yang et al., 1332 patient·es présentant une CMH apicale confirmée par IRM ont fait l’objet d’une analyse rétrospective. Un faible nombre de patient·es (2,3%) présentaient un anévrisme apical, mais 65% des anévrismes apicaux n’ont pas été diagnostiqués à l’échocardiographie.12 En utilisant des analyses de strain, notamment le «strain longitudinal global» (GLS), la contractilité globale et régionale du myocarde est déterminée avec davantage de précision. Si la fonction systolique du ventricule gauche est encore préservée, le GLS peut déjà être réduit. De cette manière, les lésions myocardiques peuvent être détectées plus tôt. De même, un GLS anormal est associé à une augmentation des événements cardiaques et des arythmies.13
Les maladies de stockage, telles que l’amylose ou la maladie de Fabry, présentent des modèles de distribution spécifiques dans l’analyse de strain. Cela peut s’avérer utile dans le cadre du diagnostic différentiel. Une réduction du strain atrial gauche dans la CMH peut en outre être un facteur prédictif de fibrillation atriale de novo.14
Importance de l’IRM cardiaque
Outre l’échocardiographie, l’utilisation de l’IRM cardiaque est recommandée pour tous les phénotypes de cardiomyopathie (recommandation de niveau 1B).1 L’IRM cardiaque combine l’avantage d’une imagerie non invasive avec une caractérisation tissulaire. Elle est particulièrement utile lorsque les conditions échocardiographiques sont mauvaises et pour évaluer des régions souvent peu visibles à l’échocardiographie, comme la paroi antérolatérale ou l’apex du ventricule gauche. La distribution du «late gadolinium enhancement» (LGE) et la présence d’une fibrose aident au diagnostic différentiel et à la pose du diagnostic.
Traitement
En principe, la CMH entraîne une insuffisance cardiaque en raison de deux mécanismes: d’une part, le phénotype obstructif et, d’autre part, la progression de la maladie avec fibrose jusqu’au stade terminal. Cependant, nombre de patient·es atteint·es de CMH restent asymptomatiques et ont une espérance de vie normale. Certain·es développent des symptômes des années après la détection d’un ECG pathologique ou d’une hypertrophie du ventricule gauche.
Comme il n’existe que peu d’études randomisées, le traitement pharmacologique repose sur une base empirique et sert avant tout à réduire les symptômes.
Traitement de la CMH obstructive
En présence d’une obstruction significative de la chambre de chasse du ventricule gauche (>50mmHg), un traitement médicamenteux doit être mis en place (Fig. 2). Sur le plan thérapeutique, l’utilisation de bêtabloquants ou de vérapamil/diltiazem est recommandée. L’inhibiteur réversible de la myosine cardiaque mavacamten est disponible depuis peu. Ces traitements peuvent être administrés seuls ou en combinaison. Les traitements invasifs disponibles sont la myectomie septale ou l’alcoolisation septale. La majorité des patient·es asymptomatiques présentant une obstruction de la chambre de chasse du ventricule gauche n’ont pas besoin de traitement.
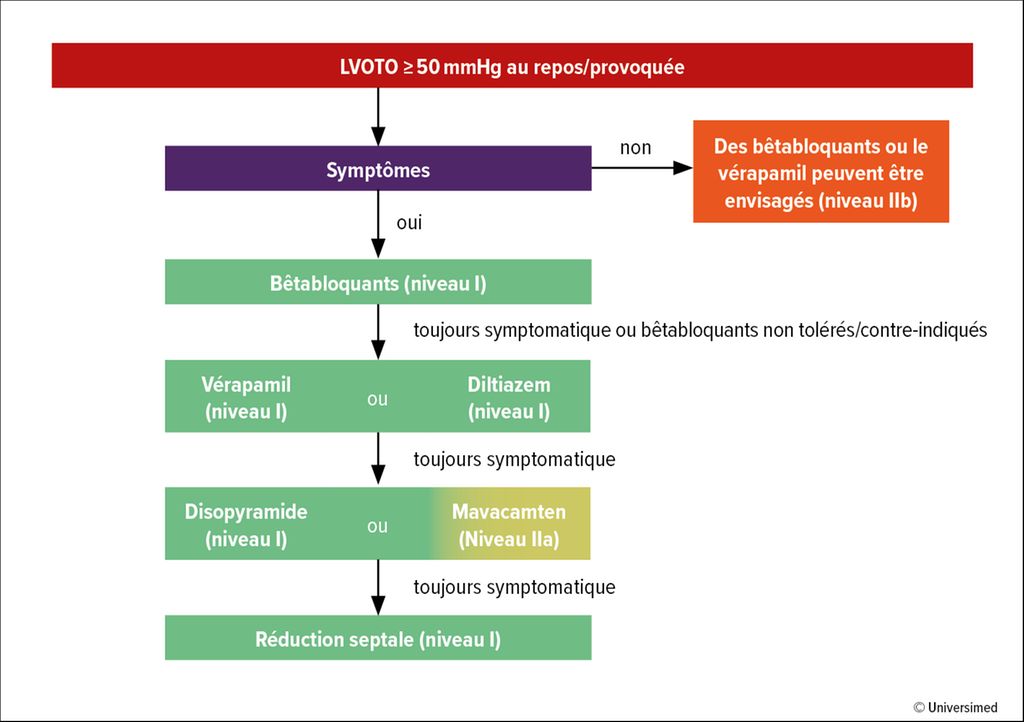
Fig.2: Décision thérapeutique chez les patient·es atteint·es de CMH obstructive symptomatiques (selon Arbelo E et al. 2023)1
Traitement de la CMH non obstructive
Conseil pratique
Si l’épaisseur de la paroi du myocarde est ≥ 15 mm, il faut envisager une cardiomyopathie hypertrophique. L’identification d’une obstruction de la chambre de chasse du ventricule gauche est importante pour le traitement.Le traitement des patient·es symptomatiques sans obstruction de la chambre de chasse du ventricule gauche (CMH non obstructive) se concentre sur le traitement des arythmies, la réduction de la pression de remplissage du ventricule gauche et le traitement de l’angine de poitrine. Les bêtabloquants ou les inhibiteurs calciques (vérapamil/diltiazem) doivent être envisagés.1 Le traitement de l’insuffisance cardiaque en cas de CMH non obstructive est recommandé conformément aux «2021 ESC Guidelines for Diagnostic and Treatment of Acute and Chronic Heart Insufficiency», par les «fantastic four» (bêtabloquants, inhibiteurs de l’ECA ou ARNI, antagonistes du récepteur des minéralocorticoïdes, inhibiteurs du SGLT2) en cas de fraction d’éjection réduite.
Évaluation d’un DAI
En ce qui concerne le risque d’arythmie maligne, une évaluation doit être menée sur la nécessité d’un défibrillateur automatique implantable (DAI). L’âge, l’épaisseur maximale de la paroi, le gradient maximal au niveau de la chambre de chasse du ventricule gauche, les antécédents familiaux de SCD, les syncopes ainsi que les arythmies jouent notamment un rôle. Le HCM Risk-SCD Calculator de l’ESC peut être utilisé comme outil utile dans la pratique clinique quotidienne.1
Littérature:
1 Arbelo E et al.: Eur Heart J 2023; 44: 3503-3626 2 Semsarian C et al.: J Am Coll Cardiol 2015; 65: 1249-54 3 Maron BJ et al.: JACC Heart Fail 2018; 6: 376-8 4 Maron BJ: N Engl J Med 2018; 379: 655-68 5 Ho CY et al.: Circulation 2018; 138: 1387-98 6 Rowin EJ, Maron BJ et al.: Am J Cardiol 2017; 120: 2256-64 7 Lorenzini M et al.: JAMA Cardiology 2020; 5: 73-80 8 Fumagalli C et al.: JAMA Cardiol 2020; 5: 65-72 9 Elliott P et al.: Eur Heart J 2014; 35: 2733-79 10 Maron MS et al.: Circulation 2006; 114: 2232-9 11 Olivotto I et al.: J Am Coll Cardiol 2008; 52: 559-66 12 Yang K et al.: Eur Heart J Cardiovasc Imaging 2020; 21: 1341-50 13 Tower-Rader A et al.: JACC Cardiovasc Imaging 2019; 12: 1930-42 14 Debonnaire P et al.:Circ Arrhythm Electrophysiol 2017; 10: e004052
Das könnte Sie auch interessieren:

Séquelles cardiopulmonaires à long terme après une infection sévère par le SARS-CoV-2
Le SARS-CoV-2 a entraîné une crise sanitaire mondiale et a posé des défis considérables aux systèmes de santé.1 Si le Covid-19 était initialement considérée comme une maladie ...

Posters et communications sélectionnés
Lors du congrès annuel de la Société Suisse d’Endocrinologie et de Diabétologie, les endocrinologues et diabétologues suisses ont fourni une vue d’ensemble de leurs nombreuses recherches ...

Analogues de l’incrétine par rapport à la chirurgie bariatrique
La question du meilleur traitement pour la perte de poids a fait l’objet d’un débat pour et contre dans le cadre du congrès 2024 de l’EASD. Alors que la facilité d’accès et la bonne ...