Vom Genotyp zum Phänotyp
Die Beziehung zwischen Genmutationen und Tumorphänotypen der chronischen lymphatischen Leukämie (CLL) ist komplex. Die Kenntnis molekularer Datenebenen und ihrer Rolle im klinischen Verlauf verbesserte das Verständnis der Genotyp-Phänotyp-Beziehungen. Dies führte schliesslich zur Entdeckung neuer prognostischer Signaturen. Eine besondere Rolle spielt dabei die Proteomik, da die Proteinfunktion u.a. die Krankheitsentwicklung massgebend beeinflusst und eine Brücke zwischen dem Genotyp und dem Phänotyp bildet.
Keypoints
-
Bei CLL-Patienten ist die detaillierte Beurteilung basierend auf genetischen Daten aktuell limitiert durch das lückenhafte Verständnis der Beziehungen zwischen Genotyp und Phänotyp.
-
Durch die Kombination verschiedener molekularer Datenebenen inklusive globaler Proteinexpressions-Daten kann die Zellfunktionalität widergespiegelt und damit die Verbindung zwischen Genotyp und Phänotyp ausgearbeitet werden.
-
Zukünftige Studien sollten neben quantitativen Protein-expressions-Daten auch strukturelle Proteindaten und deren Verbindung zu somatischen und Keimzell-Mutationen sowie Krankheitsevolution untersuchen.
Die chronische lymphatische Leukämie (CLL) ist gekennzeichnet durch genetische und klinische Heterogenität.1–5 Vielschichtige Analysen der CLL mittels Genomik, Epigenomik (z.B. DNA-Methylierung) oder Transkriptomik (RNA-Expression) sowie des Arzneimittelansprechens ex vivo und deren Verbindung mit klinischen Parametern in grossen Patientenkohorten ermöglichten Einblicke in Genotyp-Phänotyp-Beziehungen. Neue prognostische Signaturen sowie therapeutische Zielstrukturen wurden entdeckt.6–10
Die Rolle vielschichtiger molekularer Datenanalysen bei Risikoprofilen der CLL
Indem sie DNA-Methylierungsdaten mit Daten zur RNA-Expression und zum klinischen Verlauf kombinierten, konnte die Gruppe von Prof. Dan Landau DNA-Methylierungs-Treibergene definieren, die mit dem «failure-free survival» bei CLL-Patienten assoziiert sind.9 Für diese Analysen entwickelten die Autoren eine neue statistische Inferenzmethode, die es ermöglicht, Veränderungen in der DNA-Methylierung zu identifizieren, die direkt an der Tumorevolution beteiligt sind. Damit begünstigte der Algorithmus die Entdeckung epigenetischer Mechanismen, die für die Überlebensfähigkeit von CLL-Zellen verantwortlich sind.9
Mittels Multi-Omics-Faktorenanalyse (MOFA) untersuchten die Gruppen von Dr. Wolfgang Huber und Prof. Thorsten Zenz Daten der Genomik, Epigenomik, Transkriptomik, des Arzneimittelansprechens ex vivo und des klinischen Verlaufs bei 217 CLL-Patienten. Dies ermöglichte die Entdeckung einer neuen unabhängigen prognostischen Signatur für die CLL, die invers assoziiert ist mit der Lymphozyten-Verdopplungszeit und den proliferativen Antrieb erfasst.10 Diese Signatur wurde als «CLL Proliferative Drive» (CLL-PD) bezeichnet und ist eine wichtige Determinante des Krankheitsverlaufs bei CLL.10
Solche integrativen Analysen haben das biologische Verständnis der CLL vorangebracht und eine bessere Risikobeurteilung sowie die Entdeckung potenzieller neuer Behandlungsziele ermöglicht. Die Beziehung zwischen Genmutationen und Tumorphänotypen blieb jedoch aufgrund der fehlenden Dimension der Proteinexpressionteilweise unbekannt.
Proteinexpression – die Brücke zwischen somatischen Mutationen und dem Tumorphänotyp
Die Proteinfunktion bestimmt die Zellfunktionalität und hat damit wesentlichen Einfluss auf die Krankheitsentwicklung.11,12 Bis vor Kurzem basierten die Studien zur globalen Proteinexpression in der CLL auf relativ kleinen Patientenkohorten (n=6–18).13–16 Unter Verwendung der Massenspektrometrie-basierten quantitativen Proteomik konnten trotzdem Proteinsignaturen der CLL nachgewiesen werden, einschliesslich der Hochregulierung von B-Zell-Rezeptor-Signalkomponenten,13 der Dysregulation von Spleissosomproteinen,13der Herunterregulierung von Proteinkinase-C-Signalmitgliedern14 und unterschiedlichen Profilen für unmutierte und mutierte IGHV(«Immunoglobulin heavy chain»)-CLL.15,16 Die kleinen Kohorten und unterschiedlichen methodischen Ansätze führten jedoch oft zu schlecht reproduzierbaren Ergebnissen.17
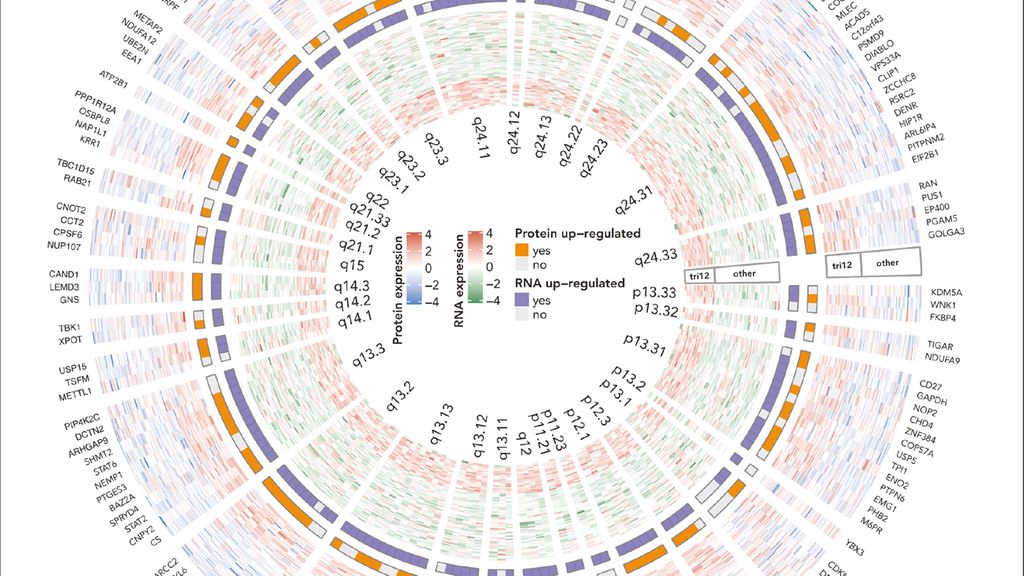
Um diesbezüglich Klärung zu schaffen, entwickelten wir eine neue datenunabhängige Massenspektrometrie(DIA-MS)-Methodik für die Analyse hämatopoetischer Zellsubpopulationen.18 Wir analysierten 117 CLL-Patientenproben auf Veränderungen der Proteinexpression und kombinierten die Resultate mit Daten der Genomik, Epigenomik, Transkriptomik, des Arzneimittelansprechens ex vivo und des klinischen Verlaufs für die gleichen Patienten.17 Trisomie 12 und der IGHV-Mutationsstatus zeigten sich als die Krankheitstreiber mit dem grössten Einfluss auf die Proteinspiegel in der CLL (1055 und 542 unterschiedlich exprimierte Proteine, FDR [«false discovery rate»] =5%). Wir untersuchten die Gen-RNA-Protein-Beziehungen für die Krankheitstreiber Trisomie 12 und Trisomie 19 und fanden unterschiedliche Grade von Gendosiseffekten. Eine stärkere Pufferung der Proteinspiegel mit hochregulierten RNA- und unveränderten Proteinwerten wurde beobachtet für Trisomie 19 im Vergleich zur Trisomie-12-CLL (p=0,0002, Kolmogorov-Smirnov-Test). Diese unterschiedlichen Grade der Proteinspiegel-Pufferung sind Beispiele für die zusätzliche Information, die im Vergleich zur Transkriptomik und Genomik durch Proteinspiegelmessungen gewonnen werden kann (Abb. 1).17
Abb. 1: Koordinierte, unkoordinierte und widersprüchliche Veränderungen der Protein- und RNA-Expression in CLL.17,23 Zirkuläres Heatmap-Diagramm mit transkriptomischen und proteomischen Daten für Gene auf Chromosom 12 von CLL-Patienten mit oder ohne Trisomie 12
Wir stellten die Hypothese auf, dass die Bildung von Proteinkomplexen dazu beiträgt, das stöchiometrische Gleichgewicht von Proteinen in der CLL aufrechtzuerhalten. Tatsächlich zeigten Chromosom-12-kodierte Proteine, von denen bekannt ist, dass sie Teil stabiler Proteinkomplexe sind, ein höheres Mass an Pufferung als andere. Nicht gepufferte Proteine unterliegen dabei einem Selektionsvorteil. Angereichert unter nicht gepufferten Proteinen in Trisomie-12-CLL waren Mitglieder des PI3K-AKT-MTOR-Signalwegs, was diesen Signalweg mit der tumortreibenden Funktion der Trisomie 12 in Verbindung bringt.17
Um die Rolle der Proteinexpression für den Tumorphänotyp zu evaluieren, untersuchten wir Proteine, die mit dem klinischen Verlauf und dem Ansprechen auf Medikamente assoziiert sind. Wir entdeckten Proteinkandidaten mit Vorhersagekraft für die Zeit zur nächsten Patientenbehandlung («timetotreatment»), ein Mass der Krankheitsaggressivität bei CLL, und identifizierten die STAT2(«signal transducer and activator of transcription 2»)-Proteinexpression als prädiktiv für das Ansprechen von CLL-Zellen ex vivo auf Kinase-Inhibitoren.17
Zusammenfassung und Schritte für die Zukunft
Die vielschichtige Analyse der CLL mittels Genomik, Epigenomik, Transkriptomik, Proteomik, des Arzneimittelansprechens ex vivo und deren Verbindung zum klinischen Verlauf hat massgebend zum Verständnis grundlegender Prinzipien beigetragen, welche die RNA- und Proteinspiegel und ihre Beziehung zu somatischen Mutationen und Tumorphänotypen der CLL bestimmen.10,17
Potenzielle neue prognostische Signaturen und Kandidaten, die eine präzisere Risikobeurteilung ermöglichen, sowie potenzielle neue therapeutische Ziele wurden entdeckt (Abb. 2). Dabei sind insbesondere Veränderungen in der Proteinexpression von entscheidender Bedeutung, da die Proteinfunktionalität die Zellfunktion bestimmt und damit die Brücke zwischen Genotyp und Phänotyp bildet.
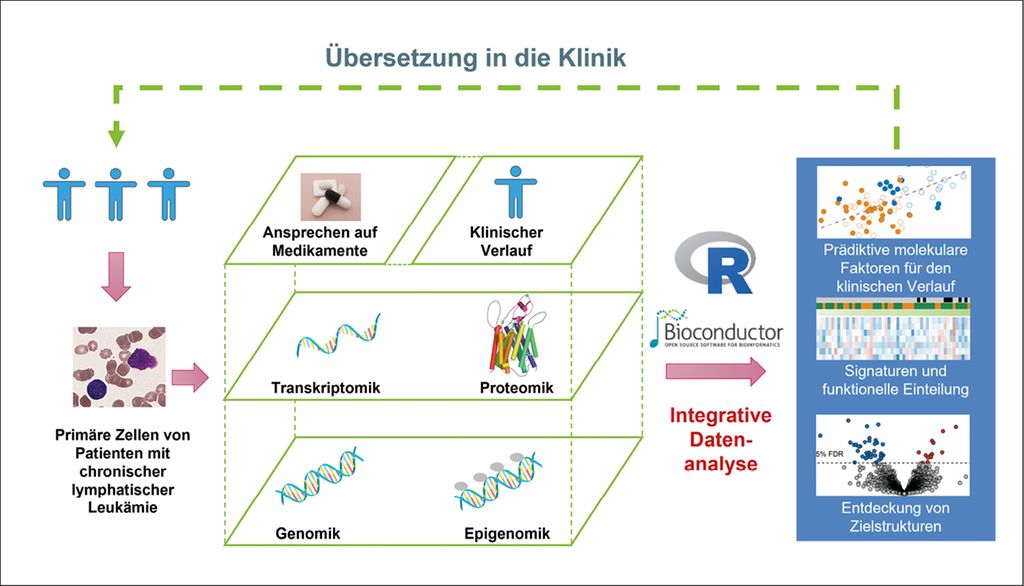
Abb. 2: Vielschichtige Analysen von CLL-Patienten ermöglichten detaillierte Einblicke in die Beziehungen zwischen Genmutationen und Tumorphänotypen. Die Abbildung wurde adaptiert von einer Darstellung von Dr. Junyan Lu. Die Proteindarstellung stammt von Meier-Abt et al.24
Die Proteinfunktionalität wird nicht nur durch die Proteinspiegel, sondern auch durch strukturelle Veränderungen beeinflusst.19,20 Viele Signalwege beruhen ausschliesslich auf strukturellen Veränderungen von Proteinen für ihre Aktivität.21,22 Zukünftige Studien sind erforderlich, um die Rolle dynamischer Veränderungen in der Proteinstruktur und ihre Beziehung zu somatischen und Keimzell-Mutationen sowie der Krankheitsevolution zu untersuchen.
Literatur:
1 Zenz T et al.: From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer 2010; 10(1): 37-50 2 Puente XS et al.: Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011; 475(7354): 101-5 3 Landau DA et al.: Mutations driving CLL and their evolution in progression and relapse. Nature 2015; 526(7574): 525-30 4 Rossi D et al.: Association between molecular lesions and specific B-cell receptor subsets in chronic lymphocytic leukemia. Blood 2013; 121(24): 4902-5 5 Mosquera Orgueira A et al.: New recurrent structural aberrations in the genome of chronic lymphocytic leukemia based on exome-sequencing data. Front Genet 2019; 10: 854 6 Ferreira PG et al.: Transcriptome characterization by RNA sequencing identifies a major molecular and clinical subdivision in chronic lymphocytic leukemia. Genome Res 2014; 24(2): 212-26 7 Dietrich S et al.: Drug-perturbation-based stratification of blood cancer. J Clin Invest 2018; 128(1): 427-45 8 Lu J et al.: Energy metabolism is co-determined by genetic variants in chronic lymphocytic leukemia and influences drug sensitivity. Haematologica 2019; 104(9): 1830-40 9 Pan H et al.: Discovery of candidate DNA methylation cancer driver genes. Cancer Discov 2021; 11(9): 2266-81 10 Lu J et al.: Multi-omics reveals clinically relevant proliferative drive associated with mTOR-MYC-OXPHOS activity in chronic lymphocytic leukemia. Nat Cancer 2021; 2(8): 853-64 11 Bludau I, Aebersold R: Proteomic and interactomic insights into the molecular basis of cell functional diversity. Nat Rev Mol Cell Biol 2020; 21(6): 327-40 12 Aebersold R, Mann M: Mass-spectrometric exploration of proteome structure and function. Nature 2016; 537(7620): 347-55 13 Johnston HE et al.: Proteomics profiling of CLL versus healthy B-cells identifies putative therapeutic targets and a subtype-independent signature of spliceosome dysregulation. Mol Cell Proteomics 2018; 17(4): 776-91 14 Diez P et al.: Multipronged functional proteomics approaches for global identification of altered cell signalling pathways in B-cell chronic lymphocytic leukaemia. Proteomics 2016; 16(8): 1193-203 15 Eagle GL et al.: Assessing technical and biological variation in SWATH-MS-based proteomic analysis of chronic lymphocytic leukaemia cells. Sci Rep 2021; 11(1): 2932 16 Eagle GL et al.: Total proteome analysis identifies migration defects as a major pathogenetic factor in immunoglobulin heavy chain variable region (IGHV)-unmutated chronic lymphocytic leukemia. Mol Cell Proteomics 2015; 14(4): 933-45 17 Meier-Abt F et al.: The protein landscape of chronic lymphocytic leukemia (CLL). Blood 2021; 138(24): 2514-25 18 Amon S et al.: Sensitive quantitative proteomics of human hematopoietic stem and progenitor cells by data-independent acquisition mass spectrometry. Mol Cell Proteomics 2019; 18(7): 1454-67 19 Ardito F et al.: The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int J Mol Med 2017; 40(2): 271-80 20 Nussinov R et al.: The underappreciated role of allostery in the cellular network. Annu Rev Biophys 2013; 42: 169-89 21 Kolch W: Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol 2005; 6(11): 827-37 22 Shaul YD, Seger R: The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta 2007; 1773(8): 1213-26 23 Thongboonkerd V: Complex systems analysis by integrative omics. Blood 2021; 138(24): 2448-50 24 Meier-Abt F, Mokrab Y: Organic anion transporting polypeptides of the OATP/SLCO superfamily: identification of new members in nonmammalian species, comparative modeling and a potential transport mode. J Membr Biol 2005; 208(3): 213-27
Das könnte Sie auch interessieren:
Erhaltungstherapie mit Atezolizumab nach adjuvanter Chemotherapie
Die zusätzliche adjuvante Gabe von Atezolizumab nach kompletter Resektion und adjuvanter Chemotherapie führte in der IMpower010-Studie zu einem signifikant verlängerten krankheitsfreien ...

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1
Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...