
Das Merkelzellkarzinom
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Das Merkelzellkarzinom (MCC) ist ein seltener neuroendokriner Tumor der Haut. Betroffen sind ältere Patienten. Die Inzidenz steigt wegen des zunehmenden Lebensalters, der erhöhten UV-Exposition und der Verwendung immunsuppressiver Medikamente. Typischerweise sind die Tumoren aggressiv, daher ist Früherkennung ebenso wichtig wie rasche Diagnose und Therapiebeginn. In diesem Übersichtsartikel werden Tumorbiologie und Immunologie, aktuelle Diagnostik und Therapiemöglichkeiten, insbesondere die Immuntherapie, diskutiert.
Keypoints
-
Das Merkelzellkarzinom ist ein aggressiver, meist schnell wachsender Tumor, dessen Diagnose schwierig sein kann. Daher gilt: schmerzlose, schnell wachsende Knoten besser biopsieren oder exzidieren als beobachten.
-
In frühen Stadien sind Operation und Strahlentherapie Standard of Care.
-
Bei fortgeschrittenen Tumoren oder im metastasierten Stadium besteht durch eine Immuntherapie mit Checkpoint-Inhibitoren, von denen in Europa derzeit nur Avelumab zugelassen ist, eine gewisse Chance auf Heilung. Probleme bereiten hier jedoch die primäre und sekundäre Resistenz. Umso mehr werden die Ergebnisse von Studien mit Checkpoint-Inhibitoren im adjuvanten und neoadjuvanten Setting erwartet.
Epidemiologie
Das MCC tritt im höheren Lebensalter auf. Das mittlere Alter bei Diagnosestellung beträgt etwa 75 bis 80 Jahre.1 50% der Tumoren treten im Kopf-Hals-Bereich auf, gefolgt von den Extremitäten mit 30%. Die Inzidenz hat seit den 1990er-Jahren zugenommen und beträgt etwa 0,13/100000 in Europa, 0,6/100000 in den USA2 und ist mit 1,6/100000/Jahr in Australien am höchsten.3 In Europa erkranken etwa 2500 Menschen pro Jahr an einem MCC.4 Männer sind etwas häufiger betroffen als Frauen.5
Pathogenese
Die Beobachtung, dass das MCC häufig an UV-exponierten Stellen der Haut auftritt und MCC-Patienten gehäuft an anderen mit UV-Licht assoziierten Krebserkrankungen (z.B. Plattenepithelkarzinome der Haut) erkranken, wurde ebenso als Hinweis auf eine pathogenetische Rolle von UV-Licht in der Genese dieser Tumoren interpretiert wie Berichte über ein UV-typisches Mutationsprofil mancher MCC6–10 oder die höhere Inzidenz in Gegenden mit hoher UV-Belastung, beispielsweise in Australien;11 daneben haben Patienten, die mit UVA oder PUVA behandelt wurden, ein höheres Risiko für ein MCC.12
2008 wurde erstmals die Assoziation einer Subgruppe von MCC mit einem neuen Polyomavirus beschrieben, welches deshalb als Merkelzellpolyomavirus (MCPyV) bezeichnet wurde.13 Weitere Untersuchungen zeigten, dass die Mehrheit der MCC (etwa 80%) in der nördlichen Hemisphäre mit diesem Virus assoziiert sind.11
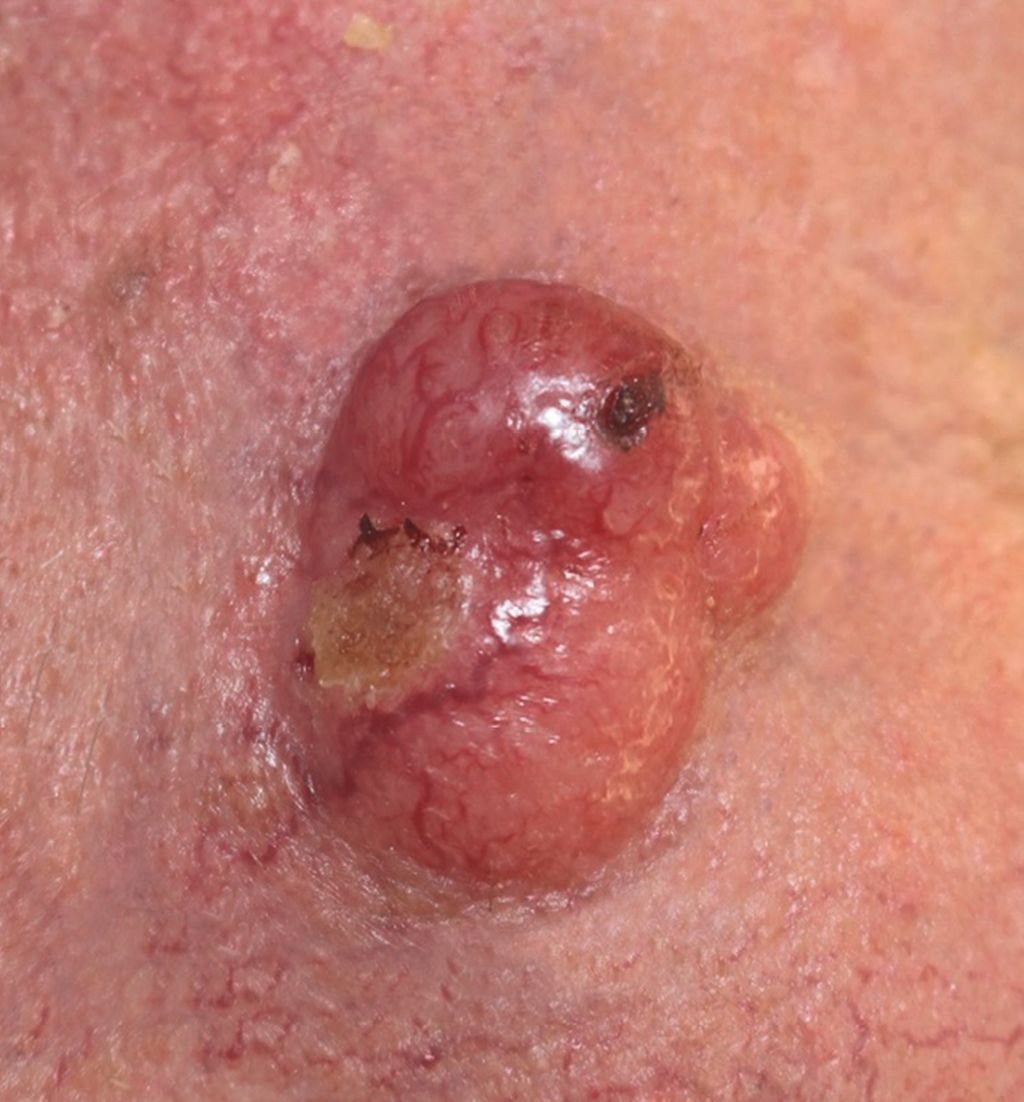
Abb. 1: Merkelzellkarzinom an der Wange: rötlicher, teils ulzerierter, kugeliger Tumor in aktinisch geschädigter Haut
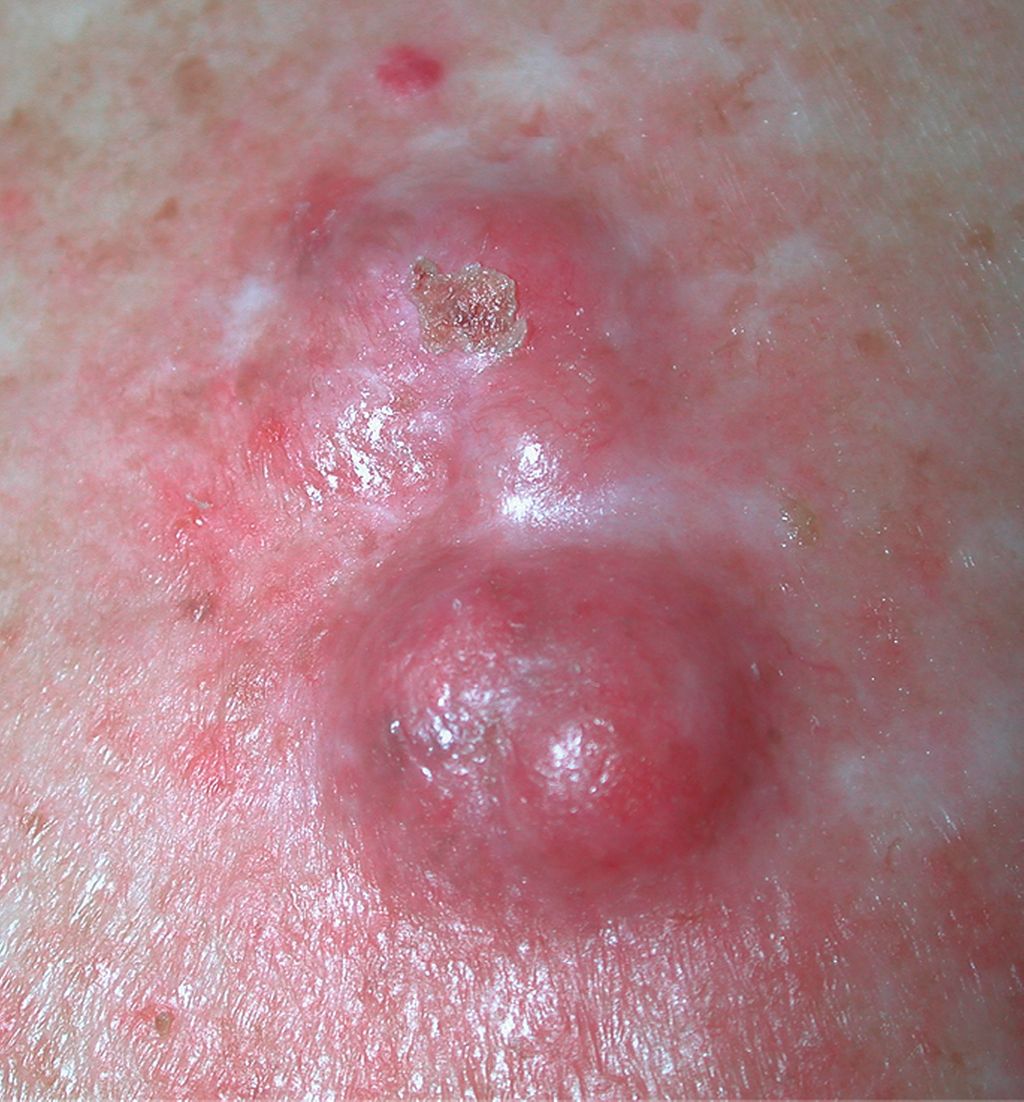
Abb. 2: MCC an der Kopfhaut: dunkelroter, kugeliger Tumor, stellenweise mit Ulzeration, ebenfalls in aktinisch geschädigter Haut
Das MCPyV wird offensichtlich bereits in der Kindheit erworben und kann auf der Haut der meisten gesunden Individuen nachgewiesen werden. Dies legt nahe, dass das MCPyV Teil des normalen Mikrobioms der Haut sein könnte.14
2010 konnten Houben et al. zeigen, dass MCPyV-positive MCC-Zelllinien von der Expression der durch MCPyV-codierten transformierenden T-Antigene LT („large T-antigen“) und ST („small T-antigen“) abhängig sind: LT und ST stellen virale Onkoproteine dar, deren Ausschaltung zum Tod der Zellen dieser MCC-Tumorzelllinien führt.15 Auch wurde gezeigt, dass das MCPyV bei Immunsupprimierten besser replizieren kann als bei Immunkompetenten.4
Zusammenfassend wird derzeit die Hypothese favorisiert, dass die Entwicklung eines MCC durch die klonale Integration des MCPyV-Genoms oder durch UV-mediierte DNA-Schädigung initiiert wird, zusätzlich könnte die UV-induzierte lokale Immunsuppression die virale Karzinogenese fördern.16
Das MCC ist ein hochimmunogener Tumor, dessen Immunogenität wahrscheinlich auf der Präsenz des MCPyV oder auf der großen UV-induzierten Mutationslast basiert.11,17 Immunsuppression, iatrogen wie auch krankheitsbedingt, stellt einen wichtigen Risikofaktor für die Entwicklung eines MCC dar. Ein besonders erhöhtes Risiko (relatives Risiko [RR] 23,8) besteht nach Organtransplantation oder bei Patienten mit hämatologischen Erkrankungen oder Aids (RR 21,6). Neben dem erhöhten Risiko haben immunsupprimierte Patienten auch eine besonders ungünstige Prognose. Sie leiden an früherem Krankheitsbeginn und höherer Mortalität des MCC.18 Zahlreiche wissenschaftliche Studien haben unser Verständnis der Pathogenese des MCC deutlich verbessert, dennoch bleibt der zelluläre Ursprung des MCC weiterhin unklar.18,19
Obwohl die Tumorzellen und normale Merkelzellen – spezialisierte Rezeptorzellen für Berührung, welche im Basalzelllager der Epidermis liegen – mehrere morphologische, immunhistochemische und ultrastrukturelle Gemeinsamkeiten haben, gibt es keine klare Evidenz für eine direkte ontogenetische Beziehung zwischen den beiden Zellen. Neben den Merkelzellen wurden epidermale oder dermale Stammzellen, stammzellenähnliche Keratinozyten und B-Zell-Vorläuferzellen als Ursprungszelle der MCC-Tumorzellen diskutiert. Weiters unterhalten Fibroblasten die MCPyV-Infektion und können somit ebenfalls zur Entwicklung des MCC beitragen. Extrakutane neuroendokrine Tumoren weisen ähnliche Merkmale wie das MCC auf.11,19
Klinik
Meistens findet sich ein hautfarbener oder rosaroter bis violetter solitärer Knoten mit glatter, glänzender Oberfläche und mäßig weicher Konsistenz, welcher in der Regel schnell wächst und rasch ulzerieren kann.
Die typischen klinischen Charakteristika können mit dem Akronym AEIOU beschrieben werden:
-
Asymptomatisch
-
Rasche Expansion
-
Immunsupprimierter und älterer („older“) Patient
-
UV-exponierte Haut
Sporadisch wird eine spontane Regression des MCC beobachtet,19 wodurch sich einzelne Fälle erklären lassen, bei denen zum Zeitpunkt der Erstdiagnose nur Metastasen, nicht aber das Primum gefunden werden.
Histologie
Histologisch gehört das MCC zu den sogenannten „Small-blue-round-cell“-Tumoren. Der Tumor besteht aus monomorphen, uniformen, kleinen, runden bis ovalen Zellen. Die Kerne dieser Zellen sind ovoid mit feinem, verstreutem Chromatinmuster („salt-and-pepper“) und groß, gelappt und hyperchromatisch. Das Zytoplasma ist spärlich. Man findet reichlich Mitosen, Einzelzellnekrosen sind häufig.20
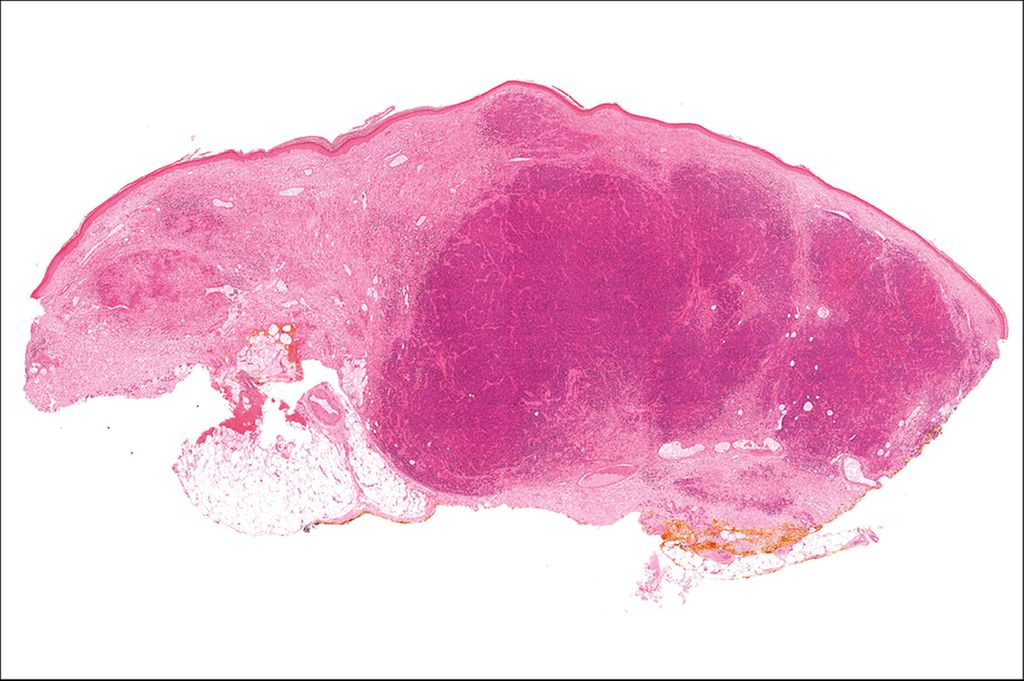
Abb. 3: Hämatoxylin/Eosin-Färbung eines Merkelzellkarzinoms in der Übersicht
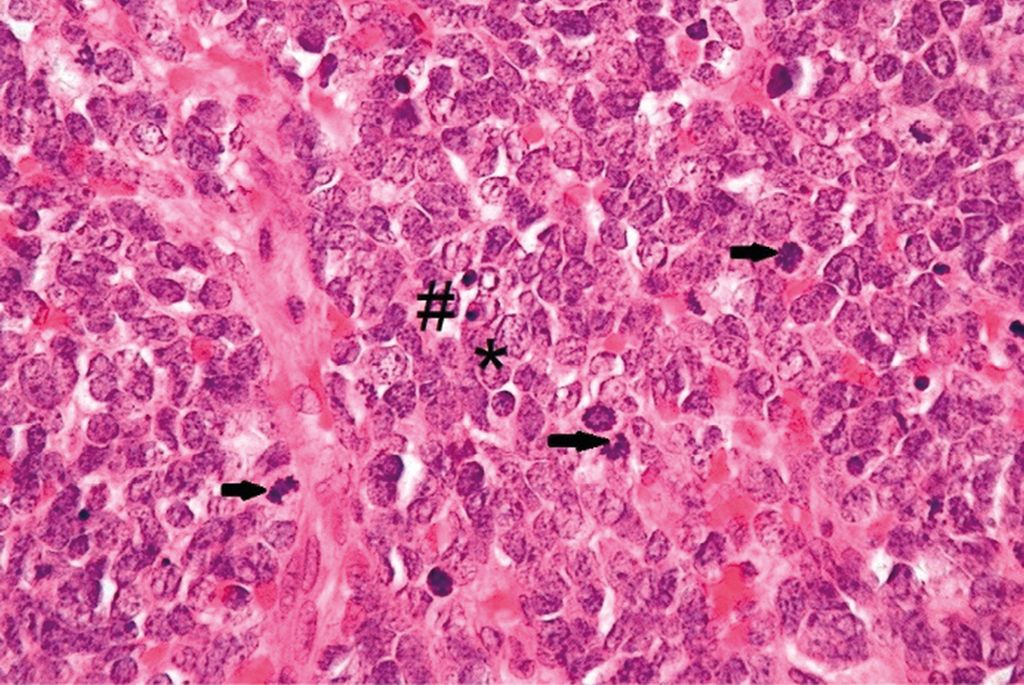
Abb. 4: Merkelzellkarzinom, Hämatoxylin/Eosin-Färbung, 600-fache Vergrößerung; * ovaläre Kerne mit „Salt-and-pepper“-Chromatin,→Mitosefiguren – teils atypisch, # pyknotische Kerne
Abbildung 3 zeigt die Übersicht eines Merkelzellkarzinoms in Hämatoxylin/Eosin-Färbung (H/E-Färbung). Man erkennt dermale und subkutane knotige Formationen eines soliden, basophilen Tumors ohne Verbindung zur Epidermis. In Abbildung 4 sieht man ein Merkelzellkarzinom (H/E-Färbung) in 600-facher Vergrößerung. Das Histologiepräparat weist dicht liegende, teils überlappende große ovaläre Kerne mit „Salt-and-pepper“-Chromatin (*), kaum abgrenzbares Zytoplasma, zahlreiche, teils atypische Mitosefiguren (→) und pyknotische Kerne (#) auf.
Ultrastrukturell findet man im Zytoplasma neuroendokrine Granula (DM 100nm), welche die MCC-Zellen definieren.
Immunhistochemisch lässt sich sowohl eine epitheliale als auch eine neuroendokrine Differenzierung nachweisen. Als epithelialer Marker gilt insbesondere Zytokeratin 20 (CK20); CK20 wird in der normalen Haut nur von Merkelzellen exprimiert und eben in den Zellen des MCC. Bei anderen neuroendokrinen Tumoren wie dem neuroendokrinen Karzinom der Lunge kommt CK20 nicht vor.
CK20 hat daher eine große diagnostische Bedeutung. Da jedoch fast 10% der MCC kein CK20 exprimieren, spielen neuroendokrine Marker wie die sehr unspezifische Neuron-spezifische Enolase (NSE), daneben Synaptophysin (nur in manchen Fällen positiv) und vor allem Chromogranin A, das Matrixprotein der neuroendokrinen Granula, eine besondere diagnostische Rolle.20
Der Nachweis des Polyomavirus entweder immunhistochemisch oder mit molekularbiologischen Methoden ist ein zusätzlicher Hinweis auf ein MCC. Die Wertigkeit des Nachweises vom MCPyV mit diesen Methoden ist jedoch für die Diagnosestellung unklar.
Diagnose
Nach genauer Inspektion des Tumors und Palpation der Lymphknotenstationen soll der Tumor vollständig exzidiert und histologisch und immunhistochemisch untersucht werden.
Mittels Bildgebung (Ultraschall, Schnittbildgebung) sollen die drainierenden Lymphknotenstationen untersucht und mittels Schnittbildgebung (CT oder 18F-FDG-PET-CT) Fernmetastasen ausgeschlossen werden. Bei unauffälligem Lymphknotenstatus wird aufgrund der hohen Zahl von okkulten Lymphknotenmetastasen – bei einem Drittel der Patienten zum Zeitpunkt der Erstdiagnose bestehend – zur besseren Einschätzung der Prognose eine Sentinel- oder Schildwächterlymphknotenbiopsie vorgenommen. Serologische Marker wie NSE oder Chromogranin A haben sich in der Diagnostik u.a. aufgrund einer hohen falsch positiven Rate als wenig hilfreich erwiesen und sollen daher nicht bestimmt werden.21,22 Potenzielle Biomarker für Verlauf und Risikoeinschätzung wie zirkulierende Tumorzellen oder Antikörper gegen virale Onkoproteine sind Gegenstand von aktuellen Untersuchungen.

Tab. 1: Klinische und histologische Differenzialdiagnosen des MCC
Stadieneinteilung
Die Stadieneinteilung erfolgt nach der aktuellen TNM-Klassifikation der AJCC (Tab. 2).

Tab. 2: Staging beim Merkelzellkarzinom nach AJCC, verkürzt
Prognose
Die Prognose des MCC hängt vom Stadium der Erkrankung ab (Tab. 2). Epidemiologische Studien zeigen regionale Unterschiede. Die 5-Jahres-Überlebensrate für Primärtumoren mit Durchmesser <2 cm liegt zwischen 66 und 75%, für Primärtumoren mit Durchmesser >2cm zwischen 42 und 52%. Für Patienten mit Fernmetastasen liegt die 5-Jahres-Überlebensrate zwischen 17 und 18%.11,23,24 Eine genauere Abschätzung der Prognose in den Stadien I und II erlaubt die Sentinel-Lymphknotenbiopsie: Die 3-Jahres-Überlebensrate in einer retrospektiven, multizentrischen Studie bei Patienten mit negativem Sentinel-Lymphknoten war 88% versus 57,2% bei Patienten mit Mikrometastasen im Sentinel-Lymphknoten.25
Behandlung
Das MCC wird mit einem adäquaten Sicherheitsabstand (SA) exzidiert. Wegen der hohen Rate an Lokalrezidiven wird die Exzision im Stadium I mit einem SA von 1cm und im Stadium II mit 2cm empfohlen.26,27 Unter Beachtung von funktionellen Ergebnissen kann in besonderen Lokalisationen (Kopf-Hals-Bereich) eine Exzision mit geringerem Sicherheitsabstand erfolgen, eventuell mit mikroskopisch kontrollierter Chirurgie und kompletter histologischer Aufarbeitung der Ränder. Eine therapeutische Lymphknotendissektion (LAD) der betroffenen Region wird bei positivem Sentinel-Lymphknoten oder Vorliegen von makroskopischen Lymphknotenmetastasen vorgeschlagen.18 Ob bei Mikrometastasen im Sentinel-Lymphknoten die therapeutische LAD zu einer Verlängerung der Überlebenszeit führt, ist derzeit noch unklar,25,28–30 daher ist eine individuelle Beratung angezeigt.
Bei Lokalrezidiven, In-Transit-Metastasen oder Lymphknotenmetastasen ist die chirurgische Resektion oder Strahlentherapie unter Berücksichtigung von operativer Belastung und Gesamtzustand zu diskutieren, die Therapie soll in kurativer Intention erfolgen. Während einzelne Organmetastasen exzidiert werden können, ist bei multiplen Metastasen eine systemische Therapie angezeigt.18
Strahlentherapie
Das MCC ist ein strahlensensibler Tumor. Die strahlentherapeutische Behandlung sollte in allen verschiedenen Stadien der Tumorerkrankung in Betracht gezogen werden18 und kann als adjuvante Maßnahme nach Tumorresektion, als primär kurativ intendierte Therapie bei Inoperabilität oder zur Palliation bei fortgeschrittener Tumorerkrankung eingesetzt werden.31,32 Wegen des hohen Risikos eines Lokalrezidivs auch nach kompletter Resektion sollte eine adjuvante Strahlentherapie des Tumorbettes mit einer Gesamtdosis von 50 Gray erfolgen. Auf diese Maßnahme kann bei Vorliegen von prognostisch günstigen Faktoren wie Tumordurchmesser unter 1cm verzichtet werden (nicht einheitlich empfohlen).24,33
Systemische Therapie
Checkpoint-Inhibitoren: neuer Goldstandard
Aufgrund der Immunogenität des MCC hat die Therapie mit Immuncheckpoint-Inhibitoren eine ganz besondere Bedeutung. Bei 10% der Patienten besteht bereits bei Erstdiagnose eine Fernmetastasierung, bevorzugt in Knochen, Gehirn, Lunge und Leber. Das Vorliegen von Fernmetastasen oder eine lokal fortgeschrittene Erkrankung, die nicht durch operative oder strahlentherapeutische Interventionen kontrolliert werden kann, stellt eine Indikation zur Systemtherapie dar. Die Immuntherapie mittels Checkpoint-Blockade (PD-1/PD-L1-Blockade) stellt den neuen „standard of care“ dar. Der PD1-PD-L1-Signalweg nimmt eine Schlüsselrolle in der Krebsbehandlung ein. Durch gezielte Blockade inhibitorischer Moleküle auf T-Zellen (PD-1) oder auf Tumorzellen (PD-L1) mit monoklonalen Antikörpern kommt es zur Reaktivierung der Immunantwort gegen verschiedene Krebsarten.
Die ersten Studien zur Therapie des metastasierten MCC mit den Anti-PD-1-Antikörpern Pembrolizumab und Nivolumab zeigten vielversprechende Ergebnisse: Pembrolizumab wies eine Ansprechrate von 56% auf, mit Nivolumab erreichten bei zuvor unbehandelten Patienten 73% ein objektives Ansprechen, bei vorbehandelten Patienten 50%. Nach einem Jahr waren drei Viertel der Remissionen noch andauernd.34
In der Phase-II-Studie JAVELIN Merkel 200 wurden 88 mit Chemotherapie vorbehandelte Patienten mit metastasiertem MCC mit dem Anti-PD-L1-Antikörper Avelumab 10mg/kg alle 2 Wochen behandelt. Hier erreichten 31,8% ein objektives Ansprechen, wobei die Ansprechrate für Patienten, die nur eine Vortherapie erhalten hatten, mit 40% höher lag als bei Patienten mit zwei oder mehr Vortherapien.4,35
Nach 12 Monaten lag das komplette Ansprechen bei 11,4% und das partielle Ansprechen bei 21,6%. Bei 71% der Responder hielt das Ansprechen mindestens 12 Monate an. Das mediane Gesamtüberleben lag bei 12,9 Monaten. 40% der Teilnehmer lebten noch nach 18 Monaten, 29% der Gesamtgruppe progressionsfrei. Avelumab war im Allgemeinen gut verträglich, in der Studie gab es keine Grad-4-Nebenwirkungen oder Todesfälle.36
Bei nachfolgender Erweiterung in der Erstlinie lag die Ansprechrate sogar bei 62%. 16 von 18 Ansprechern zeigten eine Remission bereits im ersten Staging nach sechs Wochen.36 Aufgrund dieser Daten wurde Avelumab von der FDA im März 2017 für das metastasierte MCC zugelassen, die Zulassung von der EMA folgte im September des gleichen Jahres. Die aktuell empfohlene Dosierung lautet nun 800mg Avelumab „flat dose“ alle 2 Wochen.
Auf Basis der KEYNOTE-017-Studie wurde ein Jahr später, im Dezember 2018, der PD-1-Antikörper Pembrolizumab für die Behandlung des metastasierten MCC von der FDA zugelassen.34 Mit Pembrolizumab 2mg/kg KG alle 3 Wochen konnte eine Ansprechrate (ORR; „overall response rate“) von 56% erzielt werden (komplette Remission bei 24%, partielle Remission bei 32% der Patienten). Das 2-Jahres-Überleben betrug 68,7%. Trotz dieser guten Datenlage mit dauerhafter Krankheitskontrolle und akzeptablem Sicherheitsprofil besteht für Pembrolizumab in der Indikation MCC derzeit keine EMA-Zulassung.
In diesen Studien war das Ansprechen vom Virusstatus und der PD-L1-Expression unabhängig. Verschiedene Checkpoint-Inhibitoren werden derzeit auch in der adjuvanten Situation getestet. Die Ergebnisse der prospektiven Studien müssen abgewartet werden. Erste Berichte zeigten die therapeutischen Effekte von Nivolumab und Avelumab optimistisch.37,38 Auch Pembrolizumab wird im adjuvanten Setting untersucht (STAMP Trial: Surgically Treated Adjuvant Merkel Cell Carcinoma with Pembrolizumab Stage I-IIIB MCC39).
Negative Ergebnisse erbrachte hingegen die adjuvante Behandlung mit Ipilimumab: keine Reduktion der Krankheitsprogression, jedoch deutliche Toxizität. Die DeCOG-Phase-II-Studie „ADMEC“ wurde vorzeitig beendet.40
Auch in der neoadjuvanten Situation werden PD-1-Antikörper untersucht, also mit dem Ziel einer Reduktion von Tumormasse bzw. einem Erreichen der Operabilität: In der Studie CheckMate 358 bei MCC ST IIA bis IV erhielten 39 Patienten 2 Gaben Nivolumab 240mg an Tag 1 und Tag 15, gefolgt von Resektion der Metastasen an Tag 29. Erstaunlicherweise war die Indikator-Metastase bei 47,2% verschwunden (bei 47,2% eine komplette pathologische Response). Diese Response war unabhängig von der Tumormutationslast, dem PD-L1-Status und dem MCPyV-Status.41
Dauer der Therapieantwort, Resistenz und Rechallenge
In einer retrospektiven, multizentrischen Studie der DeCOG zeigten sich raschere Rezidive nach Absetzen der Immuntherapie als beim Melanom. Die Antwort schien also nicht so lange anhaltend wie beim Melanom,44 12 von 20 Patienten erlitten einen Rückfall. Allerdings erhielten 8 dieser 12 Patienten eine Reinduktion einer Therapie mit Immuncheckpoint-Inhibitoren und bei 4 von diesen 8 Patienten konnte erneut ein Ansprechen erzielt werden. Trotz guten Erfolgs entwickeln viele Patienten von vornherein keine Antwort oder nach anfänglichem Ansprechen eine Resistenz gegen die Immuntherapie (primäre oder sekundäre Resistenz). In der Situation einer primären oder sekundären Resistenz erscheinen sequenzielle Checkpoint-Inhibitor-Therapien oder Kombinationen vielversprechend. Mehrere Studien kamen hier allerdings zu unterschiedlichen Ergebnissen:
In einer retrospektiven Studie von Glutsch et al. in Deutschland an 3 Zentren zeigten 3 von 5 Patienten mit Progress unter Avelumab (10mg/kg KG als Erstlinientherapie) eine Antwort auf Ipilimumab/Nivolumab, möglicherweise also eine vielversprechende Zweit- oder sogar Drittlinientherapie bei Patienten mit Anti-PD-L1- refraktärem MCC.45 Diese ersten Ergebnisse wurden weiter untermauert: Von derselben Gruppe wurde bei 14 Patienten eine ORR von 50% auf Ipilimumab/Nivolumab nach Resistenz auf Avelumab gezeigt (partielle oder komplette Remission). Bei einer mittleren Beobachtungszeit von 19 Monaten waren noch 63,8% der Patienten am Leben, sprich 9 von 14.46
Nicht ganz so gut sind die Daten von LoPiccolo et al.47 In einer multizentrischen retrospektiven Fallsammlung (n=13) mit Zweitlinie Checkpoint-Inhibitor-Therapie (Nivolumab, Pembrolizumab oder Avelumab) nach Progress unter zuvor erfolgter Checkpoint-Inhibitor-Therapie konnte eine ORR von 31% erzielt werden.
Noch ungünstigere Ergebnisse zeigte die Gruppe um Shalhout et al. auf der ASCO-Tagung 2022 in Chicago.48 Patienten mit Progress unter Pembrolizumab, Avelumab oder Nivolumab wurden einer Rechallenge mit Ipilimumab/Nivolumab zugeführt. Leider gab es keine komplette Remission nach RECIST-Kriterien, lediglich 23% erreichten eine stabile Erkrankung, jedoch 31% erlitten Grad-3/4-Nebenwirkungen. Das mediane progressionsfreie Überleben betrug nur 1,3 Monate, das mediane Gesamtüberleben nach Beginn der Therapie mit Ipilimumab/Nivolumab nur 4,7 Monate. Hier zeigte sich also ein limitierter Benefit von Ipilimumab/Nivolumab für Patienten mit fortgeschrittenem, Anti-PD-1/PD-L1-refraktärem MCC.
Experimentelle Ansätze bei Resistenz gegen Checkpoint-Inhibitoren
In Studien erprobt werden Therapien mit intraläsional Talimogen Laherparepvec (T-VEC), einem gentechnisch erzeugten onkolytischen abgeschwächten Herpesvirus, oder die Kombination eines onkolytischen Herpesvirus plus Nivolumab (IGNYTE-Studie49) oder zelluläre Therapien wie in der ATTAC-MCC-Studie (Phase I/II study of autologous CD8+ and CD4+ transgenic T cells expressing a high affinity MCPyV-specific TCR combined with checkpoint inhibitors and class I MHC upregulation in patients with metastatic MCC refractory to PD-1 axis blockade50), weiters gibt es Versuche mit dem MDM2-Inhibitor Navtemadlin.
Chemotherapie
Das MCC ist in der Regel ein chemosensitiver Tumor, der allerdings zu rascher Resistenzentwicklung neigt. Die verwendeten Chemotherapieschemata orientieren sich am kleinzelligen Bronchialkarzinom, die Wirksamkeit dieser Therapie wurde aber nicht in kontrollierten Studien überprüft. Zum Einsatz kommen in Mono- oder Kombinationstherapie Anthrazykline, Antimetabolite, Bleomycin, Cyclophosphamid, Etoposid, Taxane oder Platinderivate. Die Ansprechrate in einem systematischen Review von 35 Publikationen (zumeist Fallserien) lag zwischen 23 und 61% mit höherem Ansprechen in der ersten Therapielinie (53–61%) gegenüber 23–45% in der Zweitlinie.51 Die Ansprechdauer war kurz und lag in beiden Settings unter 8 Monaten, meist eher zwischen 2 und 3 Monaten,52,53 für einen positiven Einfluss auf das Gesamtüberleben liegt keine Evidenz vor.54 Wegen der hohen Toxizität insbesondere der Kombinationstherapien müssen diese Therapieformen für die teils betagten Patienten mit eingeschränkter Leber- und Nierenfunktion und Knochenmarksreserve individuell angepasst werden.
Neues zu den Biomarkern
Viele Patienten weisen MCPyV-Kapsid-Antikörper auf, diese sind daher als Verlaufsparameter ungeeignet. Antikörper gegen das MCPyV-Onkoprotein-T-Antigen hingegen finden sich selten bei Gesunden, aber bei 52% der MCC-Patienten. Seropositivität deutet auf ein niedrigeres Rezidivrisiko in multivariaten Analysen hin. Patienten mit erfolgreicher Therapie wurden nach durchschnittlich 8,4 Monaten negativ. Ein neuerlicher Anstieg wies mit 66% positivem prädiktivem Wert auf ein Rezidiv hin, während ein Titerabfall einen negativen prädiktiven Wert von 97% bedeutete. Diese Titerbestimmung ermöglicht daher eine Einteilung in Risikogruppen bzw. kann der Titer zur Verlaufskontrolle für die Überwachung des Therapieerfolgs dienen. In einer anderen Arbeit von Arroyave bedeutete Seropositivität ein signifikant besseres 2-Jahres-krankheitsfreies Überleben („disease free survival“, DFS; p=0,04) und einen Trend für besseres 2-Jahres-Überleben, nicht signifikant (p=0,37).42
Weitere Erkenntnisse auf dem Gebiet potenzieller Biomarker:43
-
PD-L1-positive Tumoren sind eher auch McPyV-positiv
-
Daten zeigen einen Trend für besseres Gesamtüberleben bei PD-L1-positiven Tumoren versus PD-L1-negativen, jedoch nicht signifikant
-
Tumor Mutational Burden (TMB): Trend für besseren Therapieerfolg durch Checkpoint-Inhibitoren bzgl. progressionsfreien Überlebens und Gesamtüberlebens bei hohem TMB
-
Virusstatus zeigt keine starke Assoziation mit Effekt einer PD-(L)1-Therapie
Nachsorge
Zur Nachsorge von Patienten mit MCC gibt es keine publizierten, wissenschaftlich gesicherten Studiendaten. Wegen des erhöhten Risikos für lokoregionäre Rezidive in den ersten zwei Jahren18,55 nach Erstdiagnose soll eine engmaschige Nachsorge mit Bildgebung vierteljährlich erfolgen. Danach kann auf eine Nachsorge in halbjährlichen Abständen übergegangen werden. Die Dauer der Nachsorge beläuft sich meistens auf 5 Jahre, da die meisten MCC-spezifischen Todesfälle sich innerhalb der ersten 5 Jahre nach Diagnose ereignen.18,55
Literatur:
1 Zaar O et al: Merkel cell carcinoma incidence is increasing in Sweden. J Eur Dermatol Venereol 2016; 30(10): 1708-13 2 Soltani AM et al.: Merkel cell carcinoma of the hand and upper extremity: current trends and outcomes J Plast Reconstr Aesthet Surg 2014: 67(3): e71-e7 3 Youlden DR et al.: Incidence and survival for Merkel cell carcinoma in Queensland, Australia, 1993-2010. JAMA Dermatol 2014; 150(8): 864-72 4 Kaufman HL et al.: Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol 2016; 17(10): 1374-85 5 Stang A et al.: The association between geographic location and incidence of Merkel cell carcinoma in comparison to melanoma: An international assessment. Eur J Cancer 2018; 94: 47-60 6 Pellitteri PK et al.: Merkel cell carcinoma of the head and neck. Head Neck 2011; 34: 1346-54 7 Schadendorf D et al.: Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. Eur J Cancer 2017; 71: 53-69 8 Kaae J et al.: Merkel cell carcinoma: incidence, mortality, and risk of other cancers. J Nat Cancer Inst 2010; 102: 793-801 9 Harms PW et al.: Distinct gene expression profiles of viral- and nonviral-associated merkel cell carcinoma revealed by transcriptome analysis. J Invest Dermatol 2013; 133(4): 936-45 10 Wong SQ et al.: UV-associated mutations underlie the etiology of MCV-negative Merkel cell carcinoma. Cancer Res 2015; 75: 5228-34 11 Becker JC et al.: Merkel cell carcinoma. Nat Rev Dis Primers 2017; 3: 17077 12 Lunder EJ et al.: Merkel-cell carcinomas in patients treated with methoxsalen and ultraviolet A radiation. NEJM 1998; 339: 1247-8 13 Feng H et al.: Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008; 319(5866): 1096-100 14 Loyo M et al.: Quantitative detection of Merkel cell virus in human tissues and possible mode of transmission. Int J Cancer 2010; 126(12): 2991-6 15 Houben R et al.: Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol 2010; 84(14): 7064-72 16 Popp S et al.: UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer 2002; 99: 352-60 17 Paulson KG et al.: Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. JCO 2011: 29(12); 1539-46 18 S2k-Leitlinie Merkelzellkarzinom (MZK, MCC, neuroendokrines Karzinom der Haut) – Update 2018 https://onlinelibrary.wiley.com/doi/abs/10.1111/ddg.13841_g 19 Walsh NM, Cerroni L: Merkel cell carcinoma: A review. Cutan Pathol 2021; 48(3): 411-21 20 Kerl H et al.: Histopathologie der Haut. Springer Verlag 2003 21 Gaiser MR et al.: Evaluating blood levels of neuron specific enolase, chromogranin A, and circulating tumor cells as Merkel cell carcinoma biomarkers. Oncotarget 2015; 6(28): 26472-82 22 Isgro MA et al.: Neuron-specific enolase as a biomarker: biochemical and clinical aspects. Adv Exp Med Biol 2015: 867: 125-43 23 Fondain M et al.: Merkel cell carcinoma in France: a registries-based, comprehensive epidemiological survey. J Eur Acad Dermatol Venereol 2018; 32(8): 1292-6 24 Fields RC et al.: Ann Surgery 2011; 245: 465-75 25 Servy A et al.: Merkel cell carcinoma: value of sentinel lymph-node status and adjuvant radiation therapy. Ann Oncol 2016; 27: 914-9 26 Haymerle G et al.: Merkel cell carcinoma: Overall survival after open biopsy versus wide local excision. Head Neck 2015; 38(Suppl 1): E1014-8 27 Schwartz JL et al.: Clinicopathologic features of primary Merkel cell carcinoma: a detailed descriptive analysis of a large contemporary cohort. Dermatol Surg 2013; 39(7): 1009-16 28 Sridharan V et al.: Merkel cell carcinoma: a population analysis on survival. J Natl Compr Canc Netw 2016; 14(10): 1247-57 29 Gunarantne DA et al.: Sentinel lymph node biopsy in Merkel cell carcinoma: a 15-year institutional experience and statistical analysis of 721 reported cases. Br J Dermatol 2015; 174(2): 273-81 30 Kouzmina et al.: Positive sentinel lymph node biopsy predicts local metastases during the course of disease in Merkel cell carcinoma. J Plast Surg Hand Surg 2013; 47(2): 139-43 31 Veness et al.: Radiotherapy alone in patients with Merkel cell carcinoma: the Westmead Hospital experience of 41 patients. Austral J Dermatol 2014; 56(1): 19-24 32 Harrington et al.: Radiotherapy and conservative surgery in the locoregional management of Merkel cell carcinoma: The British Columbia Cancer Agency Experience. Ann Surg Oncol 2016; 23(2): 573-8 33 Frohm ML et al.: Recurrence and survival in patients with Merkel cell carcinoma undergoing surgery without adjuvant radiation therapy to the primary site. JAMA Dermatol 2016; 152(9): 1001-7 34 Nghiem P et al.: Durable tumor regression and overall survival in patients with advanced Merkel cell carcinoma receiving pembrolizumab as first-line therapy. J Clin Oncol 2019; 37(9): 693-702 35 Kaufman HL et al.: J Immunther Cancer 2018; 6(1): e7 36 D’Angelo S et al.: Efficacy and safety of first-line avelumab treatment in patients with stage IV metastatic Merkel cell carcinoma: a preplanned interim analysis of a clinical trial. JAMA Oncol 2018; 4(9): e180077 37 Shalhout SZ et al.: Immunotherapy for non-melanoma skin cancer. Curr Oncol Rep 2021; 23(11): 125 38 Ricco G et al.: Int J Mol 202; 22: 10629 39 https://clinicaltrials.gov/ct2/show/NCT03712605 : Testing pembrolizumab versus observation in patients with Merkel cell carcinoma after surgery, STAMP study 40 Becker JC et al.: Epidemiology, biology and therapy of Merkel cell carcinoma: conclusions from the EU project IMMOMEC. Cancer Immunol Immunother 2018; 67(3): 341-51 41 Topalian SL et al.: Neoadjuvant nivolumab for patients with resectable Merkel cell carcinoma in the CheckMate 358 Trial. J Clin Oncol 2020; 38(22): 2476-87 42 Arroyave AJ et al.: Merkel cell polyomavirus antibody titer predicts recurrence-free survival. Ann Surg Oncol 2022; 29(3): 1620-6 43 Gauci ML et al.: Diagnosis and treatment of Merkel cell carcinoma: European consensus-based interdisciplinary guideline - Update 2022. Eur J Cancer 2022; 171: 203-31 44 Stege HM et al.: Response durability after cessation of immune checkpoint inhibitors in patients with metastatic Merkel cell carcinoma: a retrospective multicenter DeCOG study. Cancer Immunol Immunother 2021; 70(11): 3313-22 45 Glutsch V et al.: Activity of ipilimumab plus nivolumab in avelumab-refractory Merkel cell carcinoma. Cancer Immunol Immunother 2021; 70: 2087-93 46 Glutsch V et al, Poster #138 ADO (Hannover) 2022 47 LoPiccolo J et al.: Rescue therapy for patients with anti-PD-1-refractory Merkel cell carcinoma: a multicenter, retrospective case series. J Immunother Cancer 2019; 7(1): 170 48 Shalhout: Poster #9521, ASCO-Tagung 2022 49 Study of RP1 monotherapy and RP-1 in combination with pembrolizumab (IGNYTE) https://clinicaltrials.gov/ct2/show/NCT03767348 50 Gene modified Immune Cells (FH-MCVA2TCR) in treating patients with metastatic or unresectable Merkel cell carcinoma. https://clinicaltrials.gov/ct2/show/NCT03747484 51 Ngiehm et al.: Systematic literature review of efficacy, safety and tolerability outcomes of chemotherapy regimens in patients with metastatic Merkel cell carcinoma. Future Oncol 2017; 13(14): 1263-79 52 Iyer JG et al.: Response rates and durability of chemotherapy among 62 patients with metastatic Merkel cell carcinoma. Cancer Med 2016; 5(9): 2294-301 53 Becker JC et al.: Evaluation of real-world treatment outcomes in patients with distant metastatic Merkel cell carcinoma following second-line chemotherapy in Europe. Oncotarget 2017; 8(45): 79731-41 54 Bichakjian CK et al.: Merkel cell carcinoma. Version 1.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2018; 16(6): 742-74 55 Zhou P et al.: In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature 2014; 506(7486): 52-7
Das könnte Sie auch interessieren:

Mycoplasma genitalium, Chlamydien, Syphilis
Sexuell übertragbare Infektionen sind weltweit im Ansteigen begriffen, was die Resistenzproblematik verschärft. Dass ein Screening asymptomatischer Personen nicht unbedingt die optimale ...
The use of ultrasonography to guide aesthetic filler injections
The use of aesthetic filler injections has been steadily increasing in recent years. Correspondingly, there has also been an increase in reported complications. Among these, vascular ...

Systemtherapie des HER2-low fortgeschrittenen Mammakarzinoms
HER2-low- und HER2-ultralow-Mammakarzinome stellen besondere Herausforderungen dar, da sie sich sowohl in ihrer Prognose als auch im Therapieansprechen von HER2-positiven und HER2-zero- ...