Neuigkeiten rund um die pulmonale Hypertonie
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Vieles hat sich in den letzten Jahren im Forschungsfeld der pulmonalen Hypertonie (PH) getan. 2022 wird ein lang ersehntes Update der Leitlinien aus dem Jahr 2015 erwartet.1 Dieser Artikel präsentiert zusammenfassend die bis dato gültigen Diagnostik-und Therapiestrategien der PH und Neuigkeiten rund um die pulmonale Zirkulation.
Keypoints
-
Zukünftig soll bereits ein mPAP von 21mmHg die Diagnose pulmonale Hypertonie (PH) bestätigen, was aber keinen Einfluss aufTherapieindikationen haben wird.
-
Die Risikoabschätzung durch Expertenzentren spielt in der Therapie der pulmonalarteriellen Hypertonie (PAH) eine wesentliche Rolle.
-
Bei einer PH mit Linksherzerkrankung wird therapeutisch primär auf die Optimierung der HFrEF bzw. HFpEF abgezielt.
-
Der Bedarf an neuen Behandlungsoptionen ist gerade für die PH bei Linksherzerkrankung bzw. beiLungenerkrankung besonders hoch.
-
Die Pulmonalisendarterektomie bleibt bei chronisch-thromboembolischer PH (CTEPH) weiterhin die wichtigste Therapieoption.
Klassifikation der pulmonalen Hypertonie
Nach dem 6. Weltsymposium für PH in Nizza 2018 (6.WSPH) blieb die bisherige Klassifikation der PH in 5 Gruppen, abgesehen von kleineren Anpassungen in den Subgruppen, unverändert.2Besonderes Augenmerk verdient die neue hämodynamische Definition der PH. Rezente Studien zeigen, dass bereits ein mittels Rechtsherzkatheter gemessener, mittlerer pulmonalarterieller Druck (mPAP) >20mmHg mit einer erhöhten Mortalität einhergeht.3,4 Diese Grenze markierte ohnehin den Normalbereich.5Die neue Leitlinie wird daher voraussichtlich den Cut-off für eine PH-Diagnose von 25mmHg auf 21mmHg senken. Das hat primär keine Bedeutung für die Therapieindikationen, weil alle aktuell zugelassenen Medikamente für Patienten mit einem mPAP≥25mmHg geprüft wurden und für alle anderen Patienten keine ausreichende Evidenz in der Wirksamkeit besteht.
Gruppe 1: pulmonalarterielle Hypertonie (PAH)
Bei der PAH, einer seltenen Form des Lungenhochdrucks, liegt häufig ein vasokonstriktives „Remodeling“ der kleinen Pulmonalarterien vor, das zu einer massiven Erhöhung des pulmonalen Gefäßwiderstandes und arteriellen Drucks im kleinen Kreislauf führt. Progrediente Dyspnoe, Leistungsknick und Rechtsherzbelastung bis hin zum Rechtsherzversagen sind die Folge. Mittlerweile sind über 10 gezielte Medikamente für die PAH zugelassen. Sie zählen zu den Endothelin-Rezeptor-Antagonisten (ERA; Ambrisentan, Bosentan, Macitentan), Phosphodiesterase-5-Hemmern (PDE-5i; Sildenafil, Tadalafil), Stimulatoren der Guanylatzyklase (sGC; Riociguat) und den Prostazyklin-Rezeptor-Agonisten (Epoprostenol, Iloprost, Treprostinil und Selexipag). In der Subgruppe der idiopathischen PAH (IPAH) reagieren ca. 10% sehr schnell und stark auf Vasodilatoren (Responder), was eine Indikation für einen hochdosierten Kalziumkanalblocker darstellt. Für die anderen PH-Patienten haben hochdosierte Kalziumkanalblocker keine längerfristigen positiven Effekte, sondern verursachen teilweise gravierende Nebenwirkungen.
Die Risikostratifizierung spielt bei der Therapieentscheidung eine tragende Rolle. Anhand von rezenten Studien wird die Prognoseeinschätzung unter laufender Therapie präzisiert, wobei die Betrachtung von 3 einfachen Parametern sehr hilfreich ist (Tab. 1).
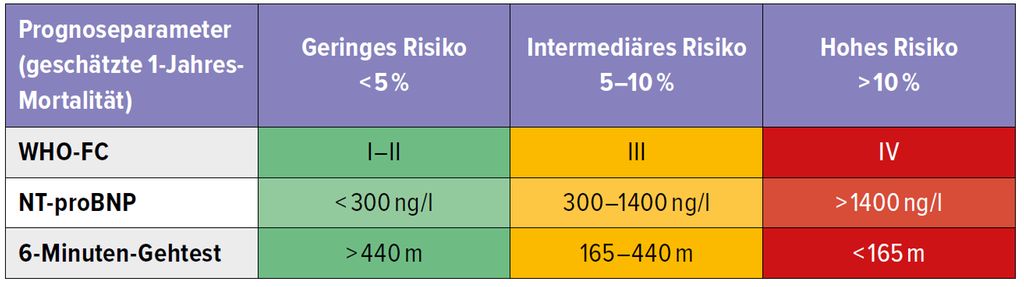
Tab. 1: Wichtige prognostische Faktoren bei PAH (modifiziert nach Galié N et al. 2019, Hoeper MM et al. 2018)2, 6
Es wird empfohlen, die Therapieentscheidungen in einem Expertenzentrum zu treffen. Die meisten Patienten werden heutzutage mit einer Kombination von gezielten PAH-Medikamenten behandelt.
Aktuell gibt es mehrere klinische Untersuchungen von neuen, gezielten Therapien der PAH. In einer Phase-III-Studie wird Sotatercept, ein Activin-Antagonist, untersucht. Es handelt sich um einen monoklonalen Antikörper, der alle 3 Wochen subkutan appliziert wird. Die entsprechende Phase-II-Studie war sehr erfolgreich auch bei PAH-Patienten, die bereits eine umfangreiche Vortherapie hatten.7
Eine weitere neue Entwicklung betrifft die Poly(ADP-Ribose)-Polymerasen (PARP), die in der onkologischen Therapie bereits als Target validiert wurden, was zu mehreren neuen Zulassungen geführt hat. Zusätzlich ist eine Vielzahl von inhalativen Medikamenten in Entwicklung. Sie zielen im Wesentlichen auf die Stimulation des Prostazyklin-Rezeptors und der löslichen Guanylatzyklase sowie auf die Inhibition des „Platelet-derivedgrowthfactor“(PDGF)-Rezeptors ab.
Gruppe 2: pulmonale Hypertonie bei Linksherzerkrankung
Diese Form der PH ist zusammen mit der PH bei Lungenerkrankungen und/oder Hypoxie die mit Abstand häufigste Ursache einer PH.8„Heart failure with reduced ejection fraction“ (HFrEF), „heart failure with preserved ejection fraction“ (HFpEF) und Klappenerkrankungen sind die wichtigsten Ursachen. Leider fehlt die Evidenz für die Behandlung solcher Erkrankungen mit PAH-Medikamenten. Die Therapie zielt auf eine optimierte Behandlung der Grunderkrankung ab.9 Manchmal ist die Abgrenzung zwischen der HFpEF und der IPAH sehr schwierig, was auch zu schwierigen Therapieentscheidungen führt. So ist zu erklären, dass im COMPERA-Register ein recht großer Teil der PAH-Patienten die typische Erscheinungsform von HFpEF-Patienten aufweist (Frauen um die 70 Jahre mit Hypertonievorgeschichte).10 Für eine PH bei Linksherzerkrankung sind die PAH-Medikamente nicht zugelassen.
Gruppe 3: pulmonale Hypertonie bei Lungenerkrankung und/oder Hypoxie
Erkrankungen der Lunge, wie COPD, interstitielle Lungenerkrankungen oder schlafbezogene Atemstörungen, sind die zweithäufigste Ursache einer PH. Die Diagnose wird bei diesen Patienten oft verkannt und häufig erst spät gestellt. Besonders überproportionale Atemnot, die trotz optimierter Therapie der Lungenerkrankung persistiert, sollte den Verdacht auf eine pulmonale Hypertonie lenken und zu einer Vorstellung in einem Expertenzentrum führen. Ob diese Patienten dann eine PAH mit einer begleitenden Lungenerkrankung (Gruppe1) oder eine PH aufgrund einer Lungenerkrankung haben (Gruppe3), ist oftmals schwer zu entscheiden und bedarf einer Vielzahl an diagnostischen Tests.11 Auch hier spiegeln sich die Abgrenzungsprobleme im COMPERA-Register wider, in dem sich eine relativ große Zahl von Patienten mit der typischen Erscheinungsform einer chronischen Lungenkrankheit findet (Männer um die 70 Jahre, viele Packungsjahre [„pack years“; py]), niedrige Kohlenmonoxid-Diffusionskapazität [DLCO]). Patienten mit einer Gruppe-3-PH, die als „schwer“ klassifiziert ist (mPAP>35mmHg oder mPAP>25mmHg und „cardiac index“ <2,0L/min/m2), können durchaus von einer gezielten PAH-Therapie profitieren.11–13 Zu diesem Thema gibt es aber noch viel Bedarf an wissenschaftlicher Aufarbeitung.
Erfreulicherweise wurden unlängst die Ergebnisse einer positiven Arzneimittelstudie (INCREASE)bei Patienten mit interstitieller Lungenkrankheit mit schwerer PH publiziert. Die Autoren zeigten in dieser multizentrischen, randomisierten,placebokontrollierten Studie, dass inhalatives Treprostinil nach 16 Wochen die körperliche Belastbarkeit signifikant verbesserte.14 In der Post-hoc-Analyse zeigte sich sogar ein günstiger Effekt auf die Lungenfibrose, obwohl das mit Vorsicht zu interpretieren ist.15
Gruppe 4: chronisch-thromboembolische Erkrankung und CTEPH
Bei der chronisch-thromboembolischen Erkrankung (CTED) kommt es aufgrund von kleinen oder großen thromboembolischen Ereignissen zu persistierenden Gefäßokklusionen und Remodeling der Pulmonalarterien. Mit Fortschreiten der pulmonalen Thromboembolien oder durch ein Remodeling der verbliebenen Gefäße kommt es zu einem Anstieg der pulmonalen Druck- und Widerstandswerte in den pathologischen Bereich, womit die chronisch-thromboembolische pulmonale Hypertonie (CTEPH) definiert wird. Oftmals ist die Unterscheidung zwischen einer akuten Pulmonalarterienembolie (PAE) und einer CTEPH eine Herausforderung. Persistieren nach einer akuten PAE die Beschwerden oder finden sich besonders hohe pulmonalarterielle Druckwerte in der Echokardiografie, sollte an eine CTEPH gedacht werden und die Patienten sollten in einem Expertenzentrum vorgestellt werden.
Prinzipiell wird bei Patienten mit CTEPH, anders als bei rezenter PAE, keine NOAK-Therapie, sondern eine dauerhafte orale Antikoagulation (OAK) mit einem Vitamin-K-Antagonisten empfohlen. Goldstandard in der Therapie ist die Pulmonalisendarterektomie (PEA). Ist diese allerdings aufgrund von zu weit peripher gelegenen Thromben oder bei nicht akzeptablem Nutzen-Risiko-Verhältnis nicht möglich, so bietet sich die Ballonpulmonalisangioplastie (BPA) als alternative Therapie an. Dabei werden die Pulmonalarterien in mehreren Sitzungen aufgedehnt.16 Es gibt aber auch medikamentöse Therapieoptionen. Für die nichtoperablen Patienten ist die orale Therapie mit Riociguat (sGC) zugelassen, und rezente Studien mit Macitentan (ERA) und Treprostinil zeigten ebenfalls positive Ergebnisse.17,18
Fazit
Die im Rahmen des 6. Weltsymposiums für PH 2018 beschlossene Änderung der PH-Definition korrigiert die Klassifikation der Patienten, was aber keine direkte Auswirkung auf die Therapie hat. Für Patientenaus der Gruppe der PAH und CTEPH mit einem mPAP>25mmHg und einem PAWP<15mmHg gibt es bereits viele zugelassene Medikamente, aber weitere Medikamente sind in Entwicklung. Die wichtigste Therapieoption für die CTEPH bleibt die Pulmonalisendarterektomie. Für die PH bei Linksherzerkrankung undPH bei Lungenerkrankung kommt leider nur selten eine PAH-Therapie infrage. Hier ist der Bedarf für die Entwicklung neuer Therapieoptionen besonders hoch.
Literatur:
1 Galiè N et al.: 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016; 37: 67-119 2 Galiè N et al.: Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J 2019; 53: 1801889 3 Douschan P et al.: Mild elevation of pulmonary arterial pressure as a predictor of mortality. Am J Respir Crit Care Med 2018; 197: 509-16 4 Assad TR et al.: Prognostic effect and longitudinal hemodynamic assessment of borderline pulmonary hypertension. JAMA Cardiol 2017; 2: 1361-8 5 Kovacs G et al.: Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J 2009; 34: 888-94 6 Hoeper MM et al.: Risk assessment in pulmonary arterial hypertension. Eur Respir J 2018; 51: 1702606 7 Humbert M et al.: Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 2021; 384: 1204-15 8 Simonneau G et al.: Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53: 1801913 9 Vachiéry JL et al.: Pulmonary hypertension due to left heart disease. Eur Respir J 2019; 53: 1801897 10 Badagliacca R et al.: Multidimensional assessment and cluster analysis for idiopathic pulmonary arterial hypertension phenotyping. J Heart Lung Transplant 2021; 40: 166-7 11 Nathan SD et al.: Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J 2019; 53: 1801914 12 Kovacs G et al.: Pulmonary vascular involvement in chronic obstructive pulmonary disease. Is there a pulmonary vascular phenotype? Am J Respir Crit Care Med 2018; 198: 1000-11 13 Bunel V et al.: Pulmonary arterial histologic lesions in patients with COPD with severe pulmonary hypertension. Chest 2019; 156: 33-44 14 Waxman A et al.: Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med 2021; 384: 325-34 15 Nathan SD et al.: Inhaled treprostinil and forced vital capacity in patients with interstitial lung disease and associated pulmonary hypertension: a post-hoc analysis of the INCREASE study. Lancet Respir Med 2021; 9: 1266-74 16 Zoppellaro G et al.: Balloon pulmonary angioplasty in patients with chronic thromboembolic pulmonary hypertension ― a systematic review and meta-analysis. Circ J 2019; 83: 1660-7 17 Ghofrani HA et al.: Macitentan for the treatment of inoperable chronic thromboembolic pulmonary hypertension (MERIT-1): results from the multicentre, phase 2, randomised, double-blind, placebo-controlled study. Lancet Respir Med 2017; 5: 785-94 18 Sadushi-Kolici R et al.: Subcutaneous treprostinil for the treatment of severe non-operable chronic thromboembolic pulmonary hypertension (CTREPH): a double-blind, phase 3, randomised controlled trial. Lancet Respir Med 2019; 7: 239-48
Das könnte Sie auch interessieren:

Chronische Atemwegserkrankungen in einem sich verändernden Klima
Die global steigenden Temperaturen und zunehmenden Hitzewellen haben einen negativen Einfluss auf die Luftqualität, vor allem in Städten. Die Atemwege und die Lunge als Eintrittspforten ...

Pathobiologie und Genetik der pulmonalen Hypertonie
Für die 7. Weltkonferenz für pulmonale Hypertonie (World Symposium on Pulmonary Hypertension; WSPH) 2024 beschäftigten sich zwei Task-Forces aus 17 internationalen Experten allein mit ...
