Periphere Nervenscheidentumoren
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Periphere Nervenscheidentumoren sind eine seltene, aber dennoch bedeutende Tumorentität, die das periphere Nervensystem betrifft. Sie können eine Vielzahl an Symptomen verursachen und eine Herausforderung in der Diagnostik und Therapie darstellen. Ein fundiertes Verständnis dieser Tumoren ist entscheidend, um eine frühzeitige Diagnose gewährleisten und adäquate Behandlungsstrategien entwickeln zu können.
Keypoints
-
Die Diagnose erfordert eine umfassende klinische Bewertung und hochauflösende bildgebende Verfahren wie High-Resolution-Ultraschall und MRT.
-
Die Therapie zielt darauf ab, den Tumor vollständig zu entfernen, ohne die umliegenden Nervenfasern zu schädigen.
-
Moderne mikrochirurgische Techniken mit intraoperativem Neuromapping ermöglichen eine faszikelschonende Enukleation mit Erhaltung der motorischen und sensiblen Nervenfunktion.
-
Die morphologische Klassifikation von BPNST in vier verschiedene Subtypen erlaubt eine differenzierte Vorgehensweise für die operative Planung und ist entscheidend für den Erfolg der Intervention.
Periphere Nervenscheidentumoren (PNST) sind seltene und im Großteil der Fälle benigne Neoplasien des peripheren Nervensystems. Diese benignen PNST (BPNST) machen 10–12% aller gutartigen Weichteiltumoren aus. Je nach histologischer Ursprungszelle wurden PNST historisch in Schwannome, Perineuriome und Neurofibrome unterteilt. Schwannome und Perineuriome bestehen aus uniformen Populationen von Schwannzellen bzw. perineuralen Zellen. Neurofibrome sind heterogene Tumoren und bestehen neben Schwannzellen auch aus Fibroblasten, Mastzellen und ggf. weiteren Zelltypen. Die WHO definiert zusätzlich Hybridtumoren, die sich anhand immunhistochemischer Kriterien unterscheiden und keiner der zuvor genannten Tumorentiäten eindeutig zugeordnet werden können. Die Ursache für PNST ist weitgehend unbekannt, wobei bestimmte Genmutationen (z.B. NF1) mit dem Auftreten von PNST assoziiert sind. PNST entarten nur selten zu malignen peripheren Nervenscheidentumoren (MPNST). Das Verhältnis benigner zu maligner PNST liegt etwa bei 9:1. MPNST stellen mit einer Inzidenz von 0,001% in der Gesamtpopulation eine seltene Tumorform dar und umfassen im Wesentlichen maligne Schwannome und Neurofibrosarkome.
Klinisch können sich PNST unterschiedlich präsentieren, wobei in der Mehrzahl der Fälle Schmerzen sowie eine tastbare Weichteilschwellung im Bereich des Tumors im Vordergrund stehen. Bei lediglich 20% der Betroffenen kommt es zu neurologischen Ausfallserscheinungen im Sinne von motorischen Defiziten, quantitativen (Hypästhesie, Anästhesie, Hyperästhesie) oder qualitativen Sensibilitätsstörungen(Dysästhesie, Parästhesie).
Diagnostik
Bei Vorliegen einer Raumforderung mit Verdacht auf einen peripheren Nerventumor ist in Hinblick auf die korrekte Diagnosestellung und die Therapieplanung eine sorgfältige Evaluation der klinischen Befunde in Zusammenschau mit hochauflösenden bildgebenden Verfahren erforderlich. Goldstandard bezüglich der radiologischen Diagnostik sind der hochauflösende Ultraschall („high resolution ultrasound“, HRUS) und die Magnetresonanztomografie (MRT). Additiv eignen sich neurophysiologische Untersuchungen zur Abschätzung der Lokalisation des Tumors und der Prognose im Verlauf sowie der präoperativen Dokumentation neurologischer Defizite.
Mittels Neurosonografie kann ein peripherer Nerventumor in den meisten Fällen sicher erkannt werden. Mit entsprechender radiologischer Expertise können Aussagen über die Lage des Tumors, die Lagebeziehung zu Faszikeln, bewegungsabhängige Kompression, Adhäsionen und Vaskularisierung bzw. Perfusion des Tumors getroffen werden. Die sonografische Textur des Tumors kann erste Hinweise auf die Tumorentität liefern. Allerdings ist die Aussagekraft des Ultraschalls vor allem in tiefer gelegenen Lokalisationen limitiert und erfordert eine ergänzende Bildgebung.
Additiv ist eine hochauflösende MRT mit i.v. Kontrastmittelgabe erforderlich, welche eine hohe Sensitivität und Spezifität in der Diagnostik von PNST und der Abgrenzung benigner von malignen Läsionen aufweist. Die Lage des Tumors, dessen Struktur und die intraläsionale Verteilung von Kontrastmittel lassen Rückschlüsse auf die Tumorentität zu. Allerdings sind mittels konventioneller MRT Faszikelstrukturen und deren genaue Lagebeziehung zum Tumor nur eingeschränkt abgrenzbar. Zur Darstellung der Nervenfasern ist die Diffusions-Tensor-Traktografie (DTT) die einzige etablierte In-vivo-Methode, siefindetvor allem in der Neurochirurgie Anwendung. Diese MRT-Technologie basiert auf der dreidimensionalen Darstellung molekularer Diffusionsbewegungen von Wassermolekülen. Da unterschiedliche Gewebe unterschiedliche Diffusionsbewegungen erlauben (Anisotropie), lassen sich dadurch Faszikel von umliegendem Gewebe abgrenzen. In der Literatur wird eine gute Korrelation der präoperativ erhobenen DTT-Befunde mit dem intraoperativen Situs bezüglich der Tumorlage beschrieben.
Da PNST großteils benigne Tumoren sind und Biopsien potenziell zu einer Läsion der Nervenstrukturen führen können, ist deren Durchführung kritisch anhand klinischer und bildgebender Kriterien abzuwägen. Leitliniengemäß kann bei fehlendem Verdacht auf Malignität aufgrund des Risikos für eine Faszikelläsion und der dadurch zu erwartenden Morbidität von einer Biopsie des Tumors Abstand genommen werden.
Therapie
Ziel der Therapie peripherer Nervenscheidentumorensind die vollständige Entfernung des Tumors und die Verbesserung der dadurch verursachten Symptomatik, ohne dabei nicht betroffene Nervenfaszikel zu verletzen und iatrogen motorische bzw. sensible Defizite zu verursachen. Historisch wurde bei Vorliegen eines Nervenscheidentumors eine En-bloc-Resektion des betroffenen Nervenabschnitts mit konsekutiver Nervenrekonstruktion durchgeführt. Da benigne PNST langsam progrediente Tumoren mit guter Prognose bei entsprechender Behandlung durch erfahrene Mikrochirurg:innen sind, steht mittlerweile jedoch die Enukleation unter Schonung der umliegenden Faszikelstrukturen im Vordergrund.
Dazu wird initial der tumortragende Nerv proximal und distal im Gesunden präpariert und dargestellt. Additiv zur morphologischen Beurteilung mittels Lupenbrille bzw. Mikroskop sollte intraoperativ ein Neuromonitoring verwendet werden. Mittels Elektrostimulation und ggf. zusätzlicher EMG-Aufzeichnung können intraoperativ am bzw. im Tumor verlaufende Faszikel identifiziert werden. Dazu ist eine enge Abstimmung bezüglich des Anästhesieverfahrens notwendig, da die motorische Reizantwort durch Muskelrelaxanzien beeinträchtigt werden kann. Mittels Elektrostimulation kann im Tumorbereich jenes Areal mit der geringsten Faszikeldichte identifiziert und aufgesucht werden, um im entsprechenden Intervall die Epineurotomie durchzuführen. Diese muss parallel zum Faszikelverlauf erfolgen, um eine Faszikelläsion zu vermeiden. Benigne PNST weisen typischerweise eine echte Kapsel und eine Pseudokapsel auf, wobei Letztere Nervenfaszikel und epineurale Gefäße beinhaltet. Mikrochirurgisch werden die Darstellung der Schicht zwischen Pseudokapsel und Tumorkapsel und die stumpfe Präparation des Tumors in der entsprechenden Schicht angestrebt. Im Rahmen dessen kann in der Regel der Ursprungsfaszikel („Parent-Faszikel“) des Tumors identifiziert werden. Neurofibrome weisen häufig mehr und kräftigere Ursprungsfaszikel auf als Schwannome. Um eine Enukleation des Tumors durchführen zu können und das Rezidivrisiko zu minimieren, müssen die Ursprungsfaszikel proximal und distal im Gesunden durchtrennt werden.
Kann der Tumor nicht schonend von den Faszikeln abpräpariert werden, wäre zur vollständigen Entfernung des Tumors die historisch etablierte En-bloc-Resektion im tumorfreien Bereich erforderlich. Dieses Vorgehen muss jedoch individuell in Abhängigkeit von der klinischen Symptomatik kritisch abgewogen und präoperativ mit den Patient:innen abgesprochen werden. Wird eine En-bloc-Resektion durchgeführt, ist im Anschluss eine Rekonstruktion indiziert, um die Integrität des Nervs wiederherzustellen. Dazu wird in der Regel ein autologes Nerventransplantat (z.B. Nervus suralis) interponiert und mikrochirurgisch koaptiert.
Morphologische Subtypen
Am Kepler Universitätsklinikum wurden im Rahmen einer retrospektiven Studie 34 BPNST morphologisch untersucht und im Wesentlichen vier unterschiedliche Tumorsubtypen identifiziert. Die Klassifikation erfolgt anhand der Relation des Tumors zu den innerhalb des betroffenen Nervs verlaufenden Faszikeln. Typ-I-Tumoren sind in Relation zu den gesunden Faszikeln innerhalb des Nervs peripher lokalisiert.
Tumoren des Typs II kommen innerhalb des Nervs so zu liegen, dass eine Aufteilung in zwei Hauptfaszikel erfolgt (Abb. 1 und 2). Liegt die Tumormasse zentral innerhalb des Nervs und dezentriert die Faszikel vollständig, wird der Tumor als Typ III klassifiziert (Abb. 3 und 4). Tumoren des Typ IV schließen die Nervenfaszikel innerhalb der Tumormasse ein, sodass keine Abgrenzung zwischen Faszikeln und Tumor erfolgen kann.
©
Kepler Universitätsklinikum
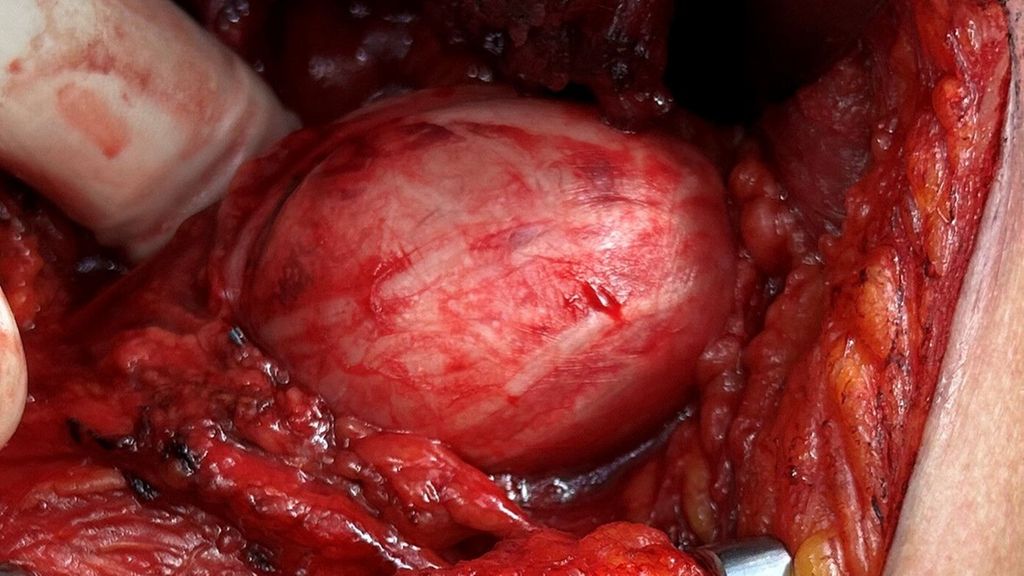
Abb. 1: Fall 1 –intraoperative Darstellung eines Schwannoms des Nervus peroneus communis rechts | 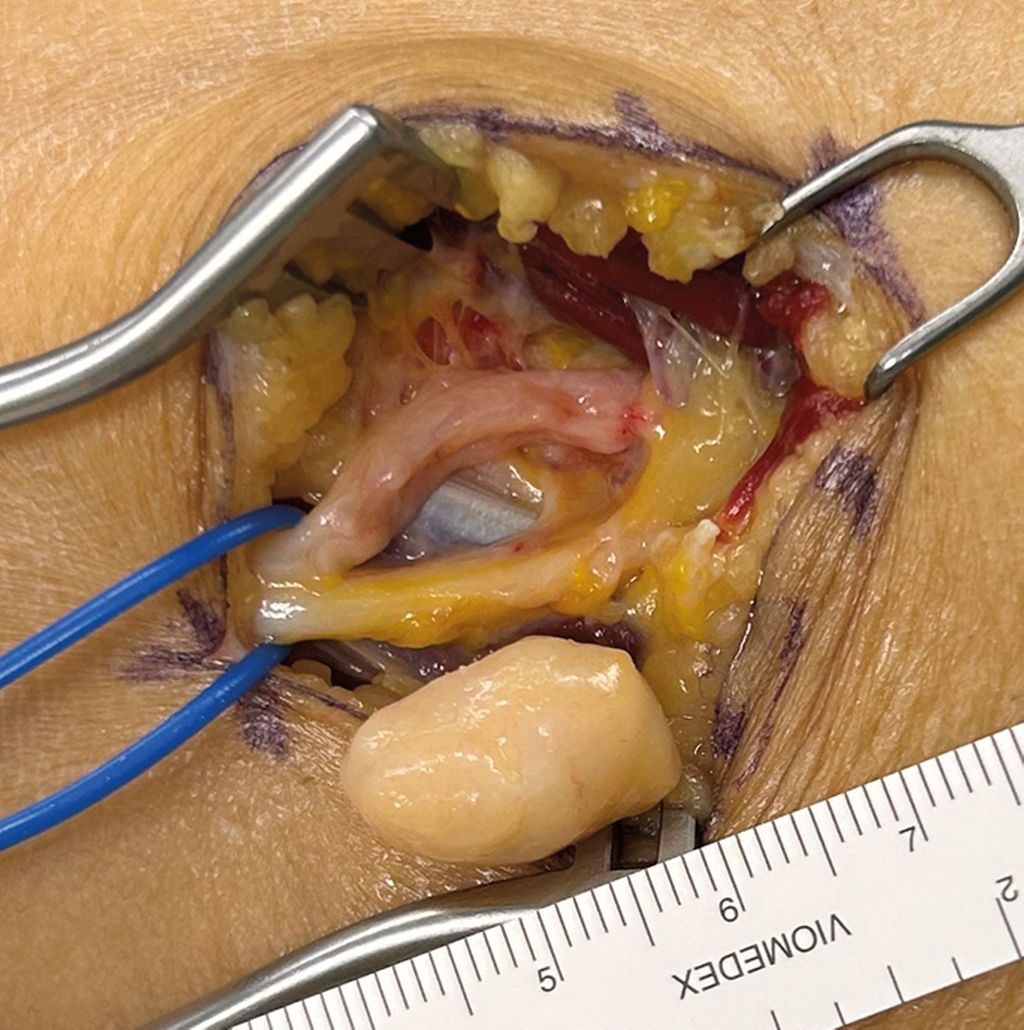
©
Kepler Universitätsklinikum
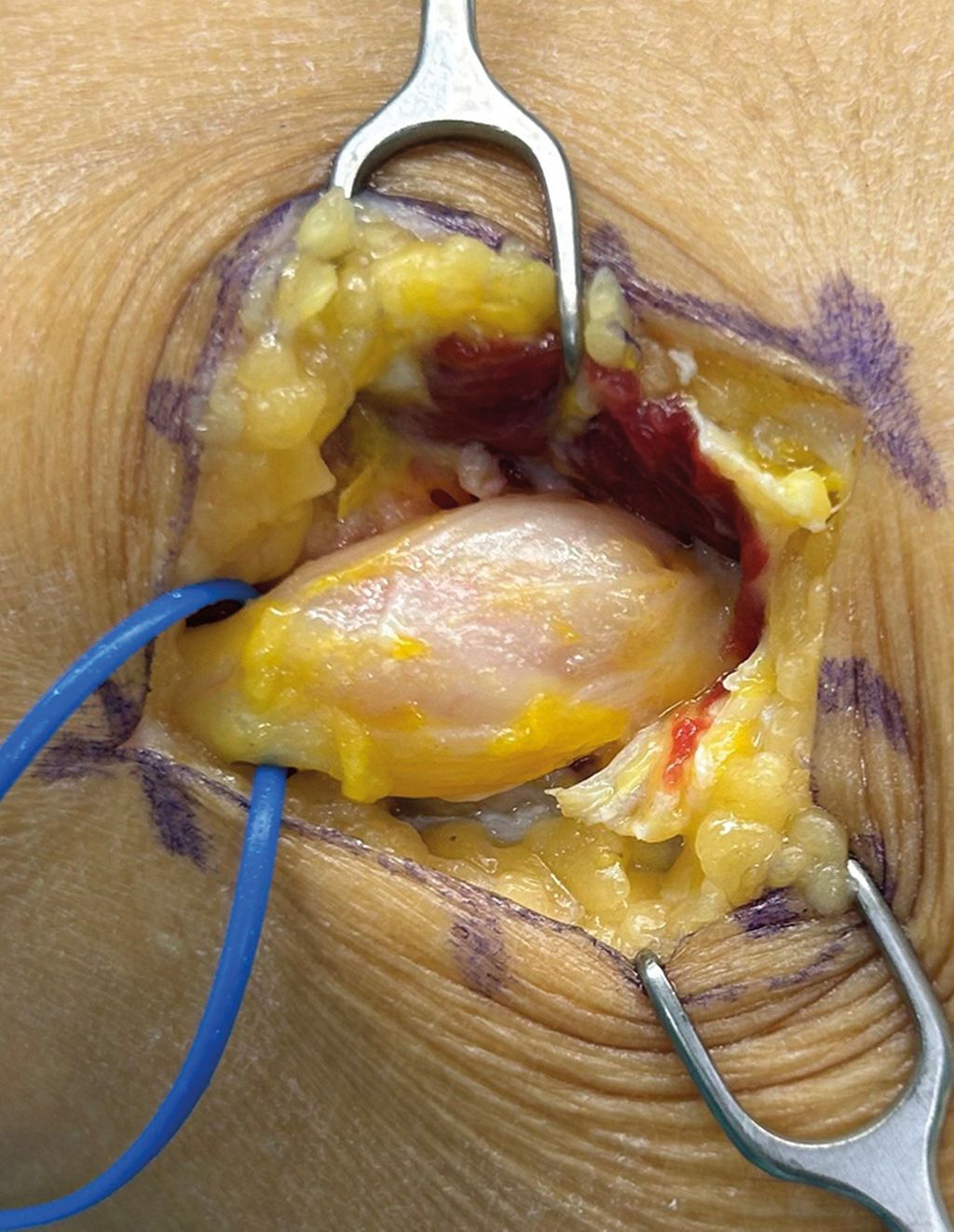
Abb. 2: Fall 1 – OP-Situs nach mikrochirurgischer Tumorexstirpation. Es zeigt sich die Aufspaltung des N. peroneus communis in zwei Faszikel, woraus sich die Klassifikation als Typ-II-Tumor ergibt |
©
Kepler Universitätsklinikum
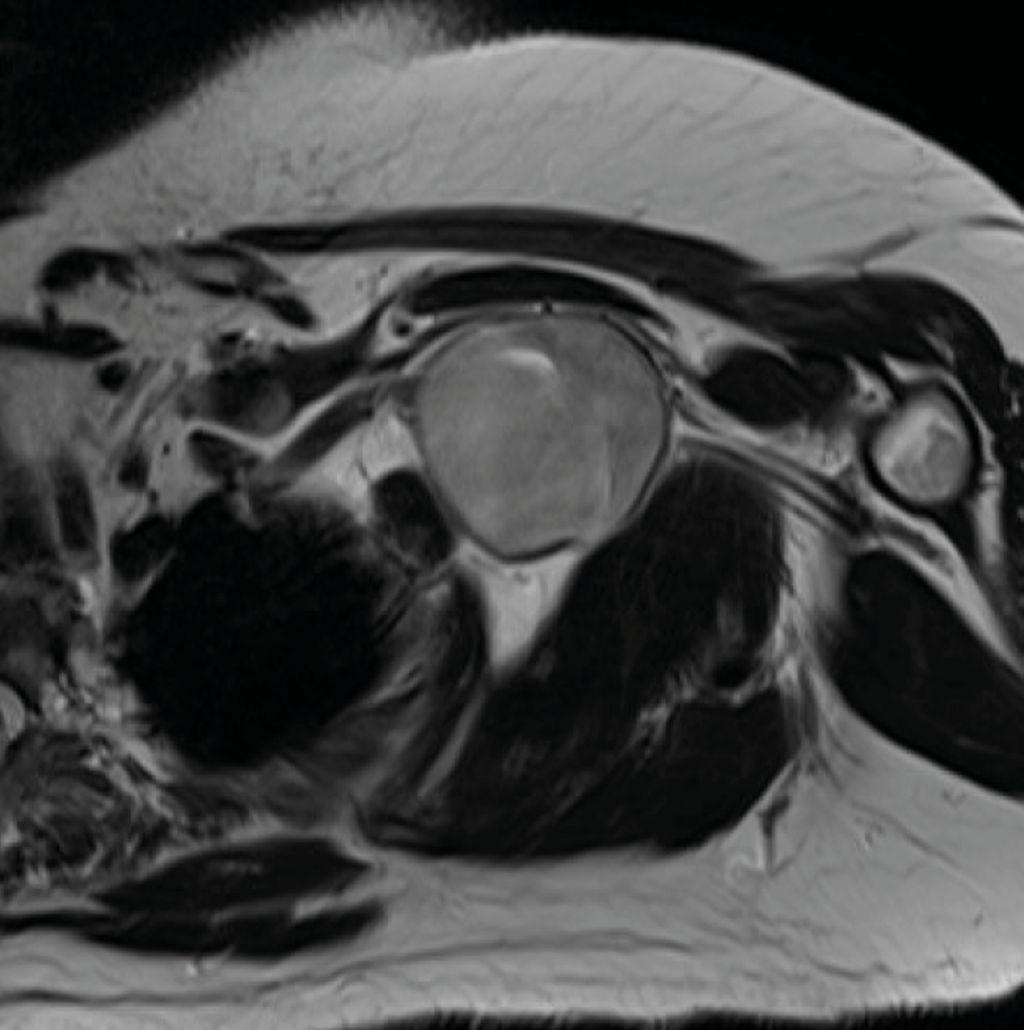
Abb. 3: Fall 2 –präoperative MRT-Darstellung eines 4,6 cm im Durchmesser haltenden, infraklavikulär, subpektoral gelegenen Nervenscheidentumors des Fasciculus posterior des Plexus brachialis mit Nahebezug zur Arteria axillaris | 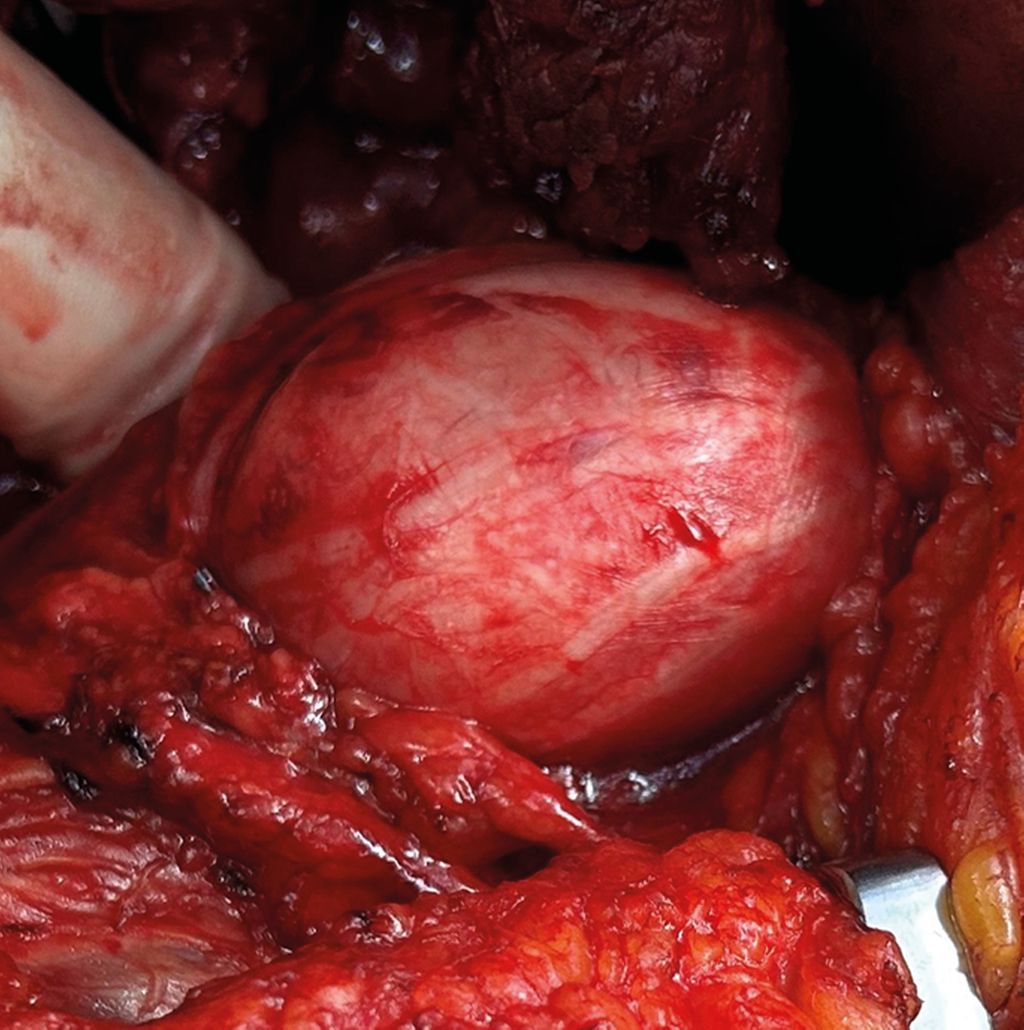
©
Kepler Universitätsklinikum
Abb. 4: Fall 2 –intraoperativer Situs mit sichtbarer Aufsplittung der Faszikel im Bereich der Tumorkapsel. Morphologische Klassifikation als Typ-III-Tumor. Die Histologie ergab ein Neurofibrom |
Mit höherem Tumorsubtyp gestaltet sich die operative Intervention komplexer, wobei bei Typ-IV-Tumoren eine En-bloc-Resektion zur vollständigen Entfernung des Tumors erforderlich wäre.
Mittels hochauflösender Bildgebung kann bereits präoperativ eine Beurteilung des Tumorsubtyps erfolgen und diese Klassifikation kann bezüglich der Operationsplanung sowie in Hinblick auf die Patient:innenaufklärung hilfreich sein.
Literatur:
1 Holzbauer M et al.: Morphological relation of peripheral nerve sheath tumors and nerve fascicles: prospective study and classification. J Clin Med 2022; 11(3) 2 Deutsche Gesellschaft für Neurochirurgie, Deutsche Gesellschaft der Plastischen, Rekonstruktiven und Ästhetischen Chirurgen, Deutsche Gesellschaft für Handchirurgie: Diagnostik und Therapie peripherer Nerventumoren. Erstversion 2022 von 4/2022, https://www.awmf.org/leitlinien/aktuelle-leitlinien.html (letzter Zugriff: 8. März 2024) 3 Kubiena H et al.: Peripheral neural sheath tumors (PNST)--what a radiologist should know. Eur J Radiol 2013; 82(1):51-5 4 Schmidt Met al.: Diffusion tensor tractography for the surgical management of peripheral nerve sheath tumors. Neurosurg Focus 2015; 39(3):E17
Das könnte Sie auch interessieren:

„Ich habe den schönsten Beruf der Welt“
Mit dem diesjährigen Jahreskongress der Österreichischen Gesellschaft für Plastische, Ästhetische und Rekonstruktive Chirurgie (ÖGPÄRC) in Salzburg hat Prim. Univ.-Doz. Dr. Rupert Koller ...

Filler sicher injizieren
Filler haben es zurzeit schwer. Gerade in der Laienpresse wird kritisch über sie berichtet (z.B. ARD/WDR: „Markt“ vom 16.10.2024). Dabei sind Filler – hier vor allem die ...
Therapie des Lymphödems
Bei der Genese des Lymphödems wird zwischen primärer und sekundärer Genese unterschieden. Derzeit werden meist nur konservative und rein symptomorientierte Therapien durchgeführt. Doch ...