Progredient fibrosierende interstitielle Lungenerkrankungen – Diagnose oder therapierelevanter Phänotyp?
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Bis vor kurzer Zeit waren die antifibrotisch wirksamen Substanzen Nintedanib und Pirfenidon der meist rasch fortschreitenden idiopathischen Lungenfibrose (IPF) vorbehalten. Studien zeigen, dass eine antifibrotische Therapie bei der progredient fibrosierenden ILD («progressive-fibrosing interstitial lung disease», PF-ILD) ebenso wirksam wie sicher ist.
Keypoints
-
Nicht nur die IPF, sondern auch andere ILD, wie die chronische exogen-allergische Alveolitis, könnten zu progredienter Lungenfibrose führen.
-
Die antifibrotischen Substanzen Nintedanib und Pirfenidon zeigten sich in aktuellen Studien auch bei progredient fibrosierenden Non-IPF-ILD wirksam.
-
Progredient fibrosierende ILD sollen – wie alle ILD – standardisiert abgeklärt und multidisziplinär in einem ILD-Board besprochen werden.
-
Kombinationstherapien aus immunsuppressiven Substanzen und Nintedanib sind nach aktuellem Wissensstand bei Subgruppen sinnvoll und sicher.
Nicht nur bei idiopathischer Lungenfibrose (IPF), sondern auch bei anderen interstitiellen Lungenerkrankungen (ILD) sind Patienten in vielen Fällen, trotz optimaler Abklärung und Therapie, von einer irreversiblen, fortschreitenden Lungenfibrose betroffen, wie z.B. bei chronischer exogen-allergischer Alveolitis oder bei einer ILD im Rahmen einer Autoimmunerkrankung.
Definition von PF-ILD
Lungenfibrose bezeichnet einen Zustand der irreversiblen Vernarbung des Lungenparenchyms, was zu einer Abnahme des Lungenvolumens (Restriktion) und zu einer Störung des Gasaustausches (Diffusionsstörung) führt. Klinisch äussert sich dies meist durch zunehmende Belastungs-, später auch Ruhedyspnoe und trockenen Reizhusten.1,2 Die klassische Form der ILD und zugleich die prognostisch ungünstigste ist die fast immer progredient fibrosierend verlaufende IPF. Hier konnten sich in den letzten Jahren die beiden antifibrotisch wirksamen Substanzen Pirfenidon und Nintedanib als Therapie der Wahl etablieren. Beide können den Abfall der Lungenfunktion signifikant verlangsamen, wenn auch nicht ganz aufhalten.2–4
Viele Arten fibrosierender interstitieller Lungenerkrankungen treten aber auch im Rahmen von chronischen pulmonalen oder systemischen Entzündungsprozessen auf, wie etwa die exogen-allergische Alveolitis (EAA), die Sarkoidose oder ILD bei Autoimmunerkrankungen, allen voran der rheumatoiden Arthritis (RA) und der systemischen Sklerose (SSC). Klassischerweise erfolgt hier primär eine Therapie der Grunderkrankung und auch der Lungenbeteiligung mit immunmodulierender/immunsuppressiver Therapie, was zu Stabilisierung oder Besserung der Lungenfunktion führen kann.5 Dennoch ist heute bekannt, dass es in ca. 30% der Non-IPF-ILD trotz umfassender Diagnostik und Therapie zu einer fortschreitenden Fibrosierung kommt.6 Für diese Gruppe wurde der Terminus «PF-ILD» eingeführt, der aber keine eigenständige Diagnose sein soll, sondern lediglich einen klinischen Phänotyp beschreibt.
Multidisziplinäre Diagnostik
Wie alle ILD sollen auch PF-ILD multidisziplinär und strukturiert abgeklärt werden. Diagnose und Therapieentscheidung sollten in einem ILD-Board besprochen werden.
Eine genaue Anamnese kann Hinweise auf die Dauer der Erkrankung und mögliche Auslöser, wie z.B. eine RA oder eine EAA, liefern. Im Hinblick auf Letztere sollte explizit die Exposition gegenüber organischen Antigenen, wie z.B. Stallarbeit, Vogelhaltung, Schimmelbelastung oder Federbettwäsche, abgefragt werden.7
Die Auskultation ist ein wichtiger Bestandteil der klinischen Untersuchung, da fibrosierende ILD zu einem charakteristischen spätinspiratorischen Knisterrasseln, der sogenannten Sklerosiphonie, führen.8 Geachtet werden soll auch auf Gelenk- oder Hautveränderungen, wie bei Kollagenosen.
Sinnvoll für die Erstabklärung – auch bei diesbezüglich asymptomatischen Patienten – ist ein Autoimmunscreening inklusive ANA, ANCA, Rheumafaktor, CCP-Antikörpern und evtl. auch Myositis-Antikörpern.1
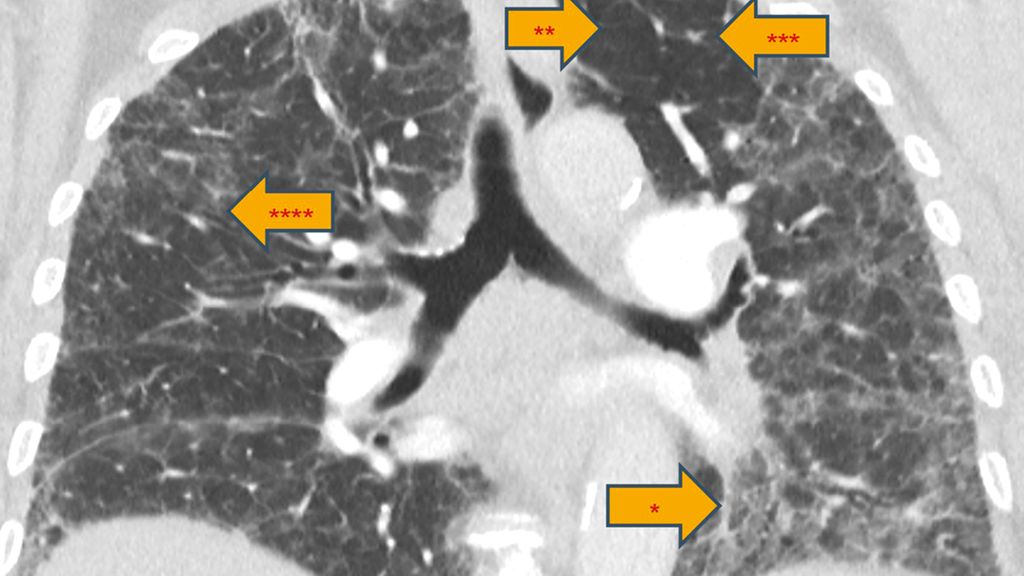
Die Diagnose einer ILD ist ohne hochauflösende Computertomografie (HRCT) nicht möglich. Hier werden verschiedene Muster beschrieben (Abb. 1 und Abb. 2). Das «Usual interstitial pneumonia»(UIP)-Muster ist klassisch für die idiopathische Lungenfibrose, tritt aber auch gehäuft bei Autoimmunerkrankungen, insbesondere bei der RA, auf. Es ist mit einer schlechteren Prognose assoziiert als beispielsweise ein «Non-specific interstitial pneumonia»-(NSIP)-Muster.9 Dennoch können verschiedene ILD mit variabler Ursache und Präsentation in der HRCT zu einer progredienten Lungenfibrose führen, beispielsweise auch die exogen-allergische Alveolitis (Abb. 3).

Abb. 1: Progredient fibrosierende ILD am Beispiel eines Patienten mit IPF im zeitlichen Verlauf mit jeweiligem 2-Jahres-Abstand: Es zeigt sich ein klassisches UIP-Muster mit peripherer subpleuraler Retikulation (*), Bildung von Honigwabenzysten («honeycombing»; **) und Traktionsbronchiektasen (***) sowie über die Zeit zunehmender Volumensreduktion der Lunge

Abb. 2: Progredient fibrosierender Verlauf einer «non-specific-interstitial-pneumonia» (NSIP) bei rheumatoider Arthritis über einen Zeitraum von 4 Jahren: Klassisch für das NSIP-Muster sind peripher und subpleural betonte retikuläre Veränderungen mit Aussparung des unmittelbaren Subpleuralraumes. Mit fortschreitender Fibrose kommt es auch hier zur Bildung von Traktionsbronchiektasen (**) und zur Volumensreduktion v.a. an der rechten Lunge

Abb. 3: Typisches Bild einer chronisch fibrosierenden exogen-allergischen Alveolitis: Klassische radiologische Befunde sind zentral und peribronchovaskulär betonte Veränderungen (*), mosaikartige Dichteunterschiede («Airtrapping» bzw. Mosaikperfusion; **) und zentrilobuläre Noduli (***). Für eine fibrosierendeILD sprechen neben retikulären Veränderungen immer auch sogenannte Traktionsbronchiektasen (****)
Lungenfunktionstestung, Diffusionskapazität und Belastungstests, wie die Spiroergometrie – oder einfacher auch der 6-Minuten-Gehtest – kommen klassisch zur Einschätzung der pulmonalen Einschränkung bei Diagnosestellung und Verlaufskontrolle zum Einsatz. Zu beachten sind auch Blutgasanalysen in Ruhe und bei Belastung, um rechtzeitig eine Sauerstofflangzeittherapie einzuleiten.
Progression bei ILD
Ob eine ILD progredient fibrosierend verläuft, lässt sich naturgemäss nur im Verlauf feststellen. Oft liegen aber schon bei ILD-Diagnose ältere Befunde vor, z.B. Thoraxröntgen oder CT bzw. Lungenfunktionsanalysen, die ein Fortschreiten der Erkrankung zeigen. In diversen klinischen Studien zu antifibrotischen Medikamenten wurden die Progression und ihr Zeitraum unterschiedlich definiert. Zusammengefasst sollte ein Zeitraum von 6 bis 24 Monaten betrachtet werden und es muss eine Verschlechterung in mehreren Untersuchungsmodalitäten vorliegen:10–13
-
Lungenfunktion: Abfall der forcierten Vitalkapazität (FVC) >5–10%
-
Diffusionskapazität: Abfall >10–15%
-
Radiologische Untersuchung: Zunahme der Fibrose in der HRCT
-
Verschlechterung der respiratorischen Beschwerden
Ebenfalls an eine Progression ist zu denken bei Verschlechterung von:
-
Leistungstests (6-Minuten-Gehtest bzw. Spiroergometrie)
-
Sauerstoffsättigung oder Sauerstoffpartialdruck in der Blutgasanalyse (in Ruhe und/oder bei Belastung)
Tritt eine Verschlechterung trotz bereits durchgeführter genauer Diagnostik und optimierter Therapie auf, wird eine Reevaluation der Diagnose und der laufenden Behandlung durchgeführt. Ergeben sich daraus keine neuen Ansätze, sollte in Anbetracht der aktuellen Datenlage eine antifibrotische Therapie angedacht werden.
Therapie von PF-ILD – aktuelle Studienlage
Angesichts der Datenlage zu den antifibrotisch wirksamen Medikamenten Pirfenidon und Nintedanib bei der fast immer progredient verlaufenden IPF lag es nahe, diese Medikamente auch für andere progressiv fibrosierende ILD einzusetzen. Nintedanib wurde zuerst erfolgreich für die ILD bei Sklerodermie erprobt und zugelassen,14 dann auch für PF-ILD auf Basis der INBUILD-Studie.10 Auch zur Substanz Pirfenidon wurden zwischenzeitlich Studien in ähnlichen Populationen publiziert, wobei sich vergleichbare Ergebnisse zeigten.11,13 Eine Zulassung besteht aktuell aber weiterhin nur für IPF.
Zu beachten ist, dass ein Teil der Patienten mit PF-ILD auch von einer immunsuppressiven Therapie profitiert, wobei Kombinationen mit Nintedanib möglich und auchwirksam sind. Insbesondere scheint dies bei Patienten mit Autoimmunerkrankungen der Fall zu sein.1,14 Zu betonen ist, dass Therapieentscheidungen bei PF-ILD immer in interdisziplinärer Zusammenarbeit mit allen beteiligten Disziplinen, insbesondere auch zwischen Pneumologie und Rheumatologie, getroffen werden sollten.
Essenziell sind auch nicht medikamentöse Massnahmen, wie pulmonale Rehabilitation, psychosoziale Betreuung, Evaluation einer Lungentransplantation (falls keine Kontraindikationen vorliegen) und bei fortgeschrittener Erkrankung auch die Etablierung einer palliativen Betreuung.1
Literatur:
1 Lang D et al.: Consensus-Statement der Österreichischen Gesellschaften für Pneumologie und Rheumatologie zur Definition, Evaluation und Therapie von progredient fibrosierenden interstitiellen Lungenerkrankungen (pfILD). Wien Klin Wochenschr 2021; 133: 23-32 2 Lederer DJ, Martinez FJ: Idiopathic pulmonary fibrosis. N Engl J Med 2018; 378: 1811-23 3 Richeldi L et al.: Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS(®) trials. Respir Med 2016; 113: 74-9 4 Noble PW et al.: Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47: 243-53 5 Tashkin DP et al.: Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med 2016; 4: 708-19 6 Wijsenbeek M, Cottin V: Spectrum of fibrotic lung diseases. N Engl J Med 2020; 3838(10): 958-68 7 Pérez ERF et al.: Diagnosis and evaluation of hypersensitivity pneumonitis: CHEST guideline and expert panel report. Chest 2021; 160: e97-e156 8 Sgalla G et al.: »Velcro-type” crackles predict specific radiologic features of fibrotic interstitial lung disease. BMC Pulm Med 2018; 18: 103 9 Kim EJ et al.: Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J 2010; 35: 1322-8 10 Flaherty KR et al.: Nintedanib in progressive fibrosing interstitial lung diseases. N Engl Med J 2019; 381: 1718-27 11 Maher TM et al.: Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med 2020; 8: 147-57 12 George PM et al.: Progressive fibrosing interstitial lung disease: clinical uncertainties, consensus recommendations, and research priorities. Lancet Respir Med 2020; 8: 925-34 13 Behr J et al.: Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med 2021; 9: 476-86 14 Distler O et al.: Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med 2019; 389: 2518-28
Das könnte Sie auch interessieren:


Chronische Atemwegserkrankungen in einem sich verändernden Klima
Die global steigenden Temperaturen und zunehmenden Hitzewellen haben einen negativen Einfluss auf die Luftqualität, vor allem in Städten. Die Atemwege und die Lunge als Eintrittspforten ...

Pathobiologie und Genetik der pulmonalen Hypertonie
Für die 7. Weltkonferenz für pulmonale Hypertonie (World Symposium on Pulmonary Hypertension; WSPH) 2024 beschäftigten sich zwei Task-Forces aus 17 internationalen Experten allein mit ...