
In a nutshell: die lymphomatoide Papulose
Selten und oft unspezifische Symptome verursachend – so präsentieren sich kutane Lymphome in der Praxis. Dieser Artikel soll einen kurzen Überblick über die am häufigsten vorkommenden T-Zell-Lymphome geben und das Krankheitsbild der lymphomatoiden Papulose anhand eines klinischen Falles vorstellen.
Keypoints
-
Das am häufigsten vorkommende kutane Lymphom ist die Mycosis fungoides.
-
73% aller kutanen Lymphome lassen sich den T-Zell-Lymphomen zuordnen, 22% den kutanen B-Zell-Lymphomen und etwa 5% weiteren seltenen Formen.
-
Bei rezidivierenden papulo-nodulären Hautveränderungen sollte an eine lymphomatoide Papulose gedacht werden.
Kutane Lymphome sind im klinischen Alltag selten und weisen eine Prävalenz von 0,1 pro 100000 Personen auf. Der Untergruppe der extranodalen Non-Hodgkin-Lymphome zugehörig, machen sie 4–5% aller diagnostizierten Lymphome aus.1
Im Unterschied zu den nodalen Lymphomen, bei welchen die entarteten Lymphozyten hauptsächlich in den Lymphknoten zu finden sind, wandern bei den primären kutanen Lymphomen die veränderten Lymphozyten in die Epidermis und Dermis ein. Beim sekundär kutanen Lymphom ist der Primärtumor ausserhalb der Haut lokalisiert (z.B. im Rahmen eines nodalen Lymphoms oder einer Leukämie) und bildet Metastasen in der Haut.
Unterteilt werden die primär kutanen Lymphome in B- und T-Zell-Lymphome, abhängig davon, ob die Erkrankung von B- oder T-Lymphozyten ausgeht. Die T-Zell-Lymphome kommen dabei verglichen mit den B-Zell-Lymphomen häufiger vor.2 Beim T-Zell Lymphom wird primär die Epidermis infiltriert (sog. Epidermotropismus), während beim B-Zell-Lymphom die Dermis sowie Subkutis betroffen ist.3
Kutanes T-Zell-Lymphom (CTCL)
Der wichtigste Vertreter der kutanen T-Zell Lymphome (CTCL) mit einem Anteil von etwa 70% stellt die Mycosis fungoides dar. Diese verläuft in drei Stadien (Ekzem-, Plaque- und Tumorstadium), wobei nur etwa 10–30% aller Krankheitsfälle das Tumorstadium erreichen.
Beim Auftreten einer Erythrodermie muss differenzialdiagnostisch an das Sézary-Syndrom gedacht werden, eine seltene, aggressive Form der CTCL.2Die Abgrenzung zwischen den beiden Krankheitsbildern erfolgt über den Nachweis von Sézary-Zellen im Blut und eine erhöhte CD4/CD8-Ratio von über 10 beim Sézary- Syndrom.3
Eine weitere wichtige Untergruppe der CTCL stellen die CD30-positiven kutanen Lymphome dar. Dazu zählen das anaplastische grosszellige Lymphom sowie die lymphomatoide Papulose, welche beide das Membranprotein CD30 überexprimieren.
Fallvorstellung
Wir berichten über einen 91-jährigen Patienten, der sich wegen eines seit etwa drei Wochen bestehenden Juckreizes und einer Hautveränderung (Knötchenbildung) am Rücken und im Flankenbereich in unserer Sprechstunde vorstellte. Hautveränderungen dieser Art waren in der Vergangenheit nicht bekannt.
Der Patient wies eine lange Liste an Vorerkrankungen auf, unter anderem eine seit 2019 vorbekannte chronische lymphatische Leukämie. Auslandsaufenthalte, Tierkontakte und das Auftreten von B-Symptomen wurden verneint. Anamnestisch war keine Atopie vorbekannt, eine Herpes-zoster-Impfung ist im Vorjahr erfolgt. Klinisch zeigten sich an der linken Flanke eine ca. 12 cm x 2cm grosse, längliche erythematöse Plaque sowie multiple wenige Millimeter messende konfluierende, erythematöse Papeln in unterschiedlichen Entwicklungsstadien (frische Papeln und Ulzerationen, teils krustig belegt). Die restliche Haut sowie die Schleimhäute präsentierten sich bland. Die Lymphknoten, die Leber und die Milz waren nicht palpabel.Zur Diagnosesicherung wurde eine histopathologische Untersuchung veranlasst. Eine Biopsie der linken Flanke bestätigte die Diagnose einer lymphomatoiden Papulose (LyP) Typ A. Molekularpathologisch wurde ein biklonales T-Zell- Rezeptor-Rearrangement gesichert. Ergänzende laborchemische Untersuchungen zeigten die im Rahmen der bereits vorbekannten CLL zu erwartenden hämatologischen Veränderungen. Zur weiteren Untersuchung wurden eine Lymphknotensonografie und ein Röntgen-Thorax veranlasst, welche keinerlei Auffälligkeiten zeigten. Wir starteten eine Lokaltherapie mit Clobetasolpropionat, worunter sich eine komplette Regression der Symptome einstellte.
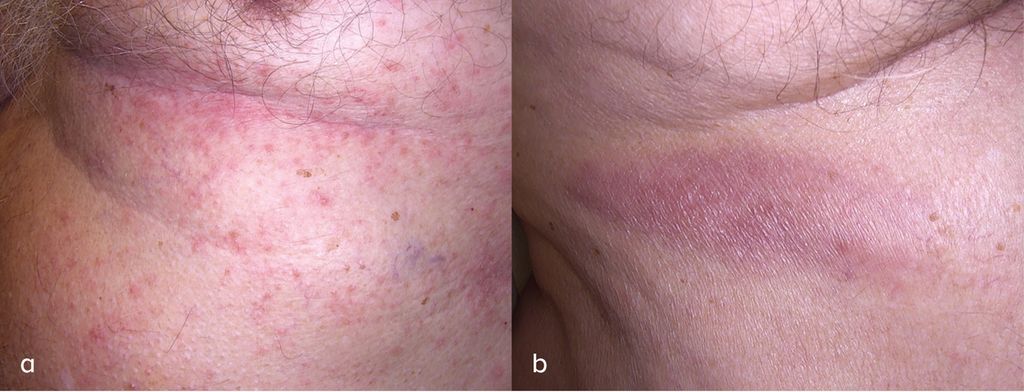
Abb. 1: a) Flanke links mit multiplen konfluierenden erythematösen Papeln; b) Flanke rechts mit ca. 12x2cm grosser länglicher erythematöser Plaque
Hintergrund lymphomatoide Papulose
Die LyP ist eine seltene, chronische auf die Haut beschränkte Erkrankung, welche durch einen jahrelangen rezidivierenden Verlauf gekennzeichnet ist. Sie macht etwa 18% aller CTCL aus.4 Der Erkrankungsgipfel liegt im 5. Lebensjahrzehnt, wobei auch ein Auftreten bei Kindern oder älteren Personen möglich ist. Tritt die Erkrankung im Kindesalter auf, ist das männliche Geschlecht häufiger betroffen, wogegen im Erwachsenenalter das weibliche Geschlecht häufiger an LyP erkrankt.5 Möglicherweise besteht eine Assoziation zu atopischen Erkrankungen. Ein gehäuftes Auftreten im Zusammenhang mit einer Infektion mit Eppstein-Barr-Viren, Herpes-Viren oder humanen T-lymphotrophen Viren konnte nicht gezeigt werden.6,7
Der Pathomechanismus beruht auf einer Überexpression des Membranproteins CD30+, welches auf den T-Lymphozyten lokalisiert ist. CD30+ aktiviert Signalwege, welche zur Proliferation und zum verlängerten Überleben von entarteten T-Lymphozyten führen.8 In >40% aller LyP-Fälle kann molekularbiologisch ein monoklonales Rearrangement von T-Zell-Rezeptor-Genen nachgewiesen werden. Untersuchungen legen nahe, dass dies ursächlich ist für das Vorliegen von T-Lymphozyten mit CD30+ Überexpression.9–12
Klinisch präsentiert sich die Erkrankung oft zeitgleich in unterschiedlichen Stadien. Zuerst zeigen sich rötliche, bräunliche oder violette, schmerzlose, oberflächenglatte Papeln begleitet von Pruritus. Die Papeln konfluieren oft innerhalb von kurzer Zeit zu roten, schmerzlosen Plaques und Knoten mit glatter Oberfläche und können im Rahmen der Abheilung krustöse Ulzerationen bilden.
Dabei lassen sich anhand des klinischen Bildes verschiedene morphologische Varianten unterscheiden, so z.B. die augmentierte, solitäre, plaqueähnliche, diffus-papulöse, tumorale, follikuläre, pustuläre, mukosale oder Borderline-LyP. Die häufigste Prädilektionsstelle ist der Rumpf, gefolgt von den Extremitäten und dem Gesicht. Eine Beteiligung der Mundhöhle ist in seltenen Fällen zu beobachten.13
Die Hautläsionen heilen oft innerhalb von 4–8 Wochen im Sinne einer Spontanremission ab und können fakultativ varioliforme Narben hinterlassen. Typisch ist ein chronisch-rezidivierender Verlauf oft über Jahre oder Jahrzehnte mit spontaner Rückbildung der Läsionen wie auch mit Episoden akuter Exazerbation («Waxing-and-Waning-Charakter»).
Nebst der Anamnese kann die Diagnose mittels Histopathologie gesichert werden. Dabei zeigen sich im Biopsat atypische CD30-positive Lymphozyten in verschiedenen Entzündungsstadien. Eine immunhistochemische Färbung sollte ergänzend dazu zur Bestätigung der CD30-positiven Lymphozyten durchgeführt werden.
Basierend auf dem histopathologischen sowie immunhistochemischen Befund wird die LyP in 6 Subtypen unterteilt, allerdings ohne prognostische Relevanz (Tab. 1).
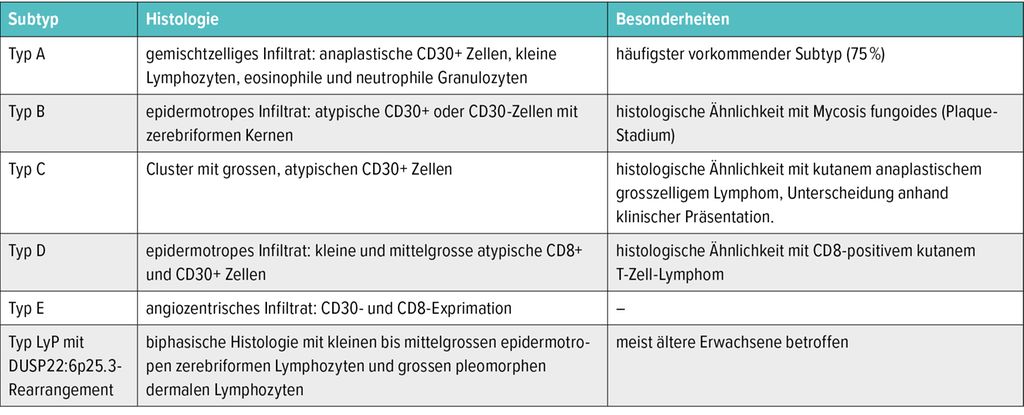
Tab. 1: Subtypen und histopathologische Merkmale der lymphomatoiden Papulose15
Ergänzend zu diesen Untersuchungen werden gemäss den Leitlinien auch eine Lymphknotensonografie sowie eine Röntgen-Thorax-Untersuchung zum Ausschluss einer systemischen Beteiligung durchgeführt.
Aufgrund des meist blanden, aber jahrelang rezidivierenden Verlaufs der Erkrankung kann bei geringem Leidensdruck und mildem Befall die «Watch-and-Wait»-Strategie angewendet werden.
Ist der Befall ausgeprägt und leiden die Patient:innen unter Juckreiz, kann die Anwendung mittelstarker bis starker Kortikosteroide sinnvoll sein. PUVA oder Schmalband-UVB stellen ebenfalls Therapieoptionen dar. Eine orale Therapie mit niedrig dosiertem Methotrexat (5–35mg/Woche) wird bei Patient:innen mit häufigen und schwerwiegenden Rezidiven empfohlen. Versagen diese Therapieoptionen erster Wahl, kann eine Therapie mit oralen Retinoiden wie Acitretin oder Bexaroten in Erwägung gezogen werden. Zeigen die Patient:innen ein hohes Rückfallrisiko oder aber zeigt sich ein Übergang der Erkrankung in ein aggressives Lymphom, kann eine zielgerichtete Therapie mittels monoklonaler Antikörper (Brentuximab) gegen CD30 durchgeführt werden.2
Etwa 20% aller an LyP-Erkrankten entwickeln im Verlauf eine andere maligne hämatologische Erkrankung. Am häufigsten treten dabei die Mycosis fungoides, das anaplastische grosszellige Lymphom sowie der Morbus Hodgkin auf. Diese Assoziation wird durch das T-Zell-Rezeptor Rearrangement erklärt, bei dem der T-Zell-Klon, der zur LyP geführt hat, auch die nachfolgenden Erkrankungen verursacht.14 Der molekulare Mechanismus, welcher für die Progression der LyP zu einer weiteren Erkrankung verantwortlich ist, ist noch nicht abschliessend geklärt.
Um eine mögliche Progression frühzeitig zu diagnostizieren, sollten PatientInnen jährlich nachkontrolliert und entsprechend sensibilisiert werden. Dabei sollte das Augenmerk auf Hautveränderungen gelegt werden, welche auf eine Mycosis fungoides bzw. andere Lymphomentitäten hindeuten, sowie anamnestisch B-Symptome erfragt werden.2
Literatur:
1 Scarisbrick JJ et al.: EORTC/UKCLG consensus statement on the management of primary cutaneous T-cell lymphomas. Br J Dermatol 2015; 173(2): 667-87 2 Dippel E et al.: SK2-Leitlinie: Kutane Lymphome (ICD10 C82-C86). Update 2021. AWMF 2021; online verfügbar unter https://register.awmf.org/assets/guidelines/032-027l_S2k_Kutane_Lymphome_2021-12.pdf (zuletzt aufgerufen am 24.9.2024) 3 Pileri A et al.: Epidemiology of cutaneous T-cell lymphomas: state of the art and a focus on the Italian Marche region. Eur J Dermatol 2023; 33: 360-7 4 Bradford PT et al.: Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood 2009; 113: 5064-73 5 Bekkenk MW et al.: Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood 2000; 95: 3653-61 6 Kadin ME et al.: Absence of Epstein-Barr viral RNA in lymphomatoid papulosis. J Pathol 1993; 170: 145-8 7 Kempf W et al.: Lymphomatoid papulosis and human herpesviruses – a PCR-based evaluation for the presence of human herpesvirus 6, 7 and 8 related herpesviruses. J Cutan Pathol 2001; 28: 29-33 8 Boi MJ et al.: CD30 in anaplastic large-cell lymphoma: a target for therapy. Clin Cancer Res 2013; 19(3): 696-703 9 Steinhoff M et al.: Single-cell analysis of CD30+ cells in lymphomatoid papulosis demonstrates a common clonal T-cell origin. Blood 2002; 100: 578-84 10 Weiss LM et al.: Clonal T-cell populations in lymphomatoid papulosis: evidence of a lymphoproliferative origin for a clinically benign disease. N Engl J Med 1986; 315: 475-9 11 Schultz JC et al.: T-cell clonality of peripheral blood lymphocytes in patients with lymphomatoid papulosis. J Am Acad Dermatol 2005; 53: 152-5 12 Humme D et al.: Dominance of nonmalignant T-cell clones and distortion of the TCR repertoire in the peripheral blood of patients with cutaneous CD30+ lymphoproliferative disorders. J Invest Dermatol 2009; 129: 89-98 13 Willemze R et al.: WHO-EORTC classification for cutaneous lymphomas. Blood 2005; 105: 3768-75 14 Gniadecki R et al.: Bone marrow precursor of extranodal T-cell lymphoma. Blood 2003; 102: 3797-9 15 Kempf W et al.: Cutaneous lymphomas: an update. Am J Dermatopathol 2017; 39(1): 37-47
Das könnte Sie auch interessieren:
Prozedurale Therapien in der Dermatologie
Prozedurale Dermatologie ist ein Teilbereich, der sich aufdiagnostische und therapeutische Methoden konzentriert, die physikalische Techniken und Eingriffe beinhalten. Dieser Artikel ...
La reconstruction mammaire immédiate par prothèses – tendances actuelles
Le cancer de la poitrine reste toujours un problème de santé publique important, 13% des femmes développant une tumeur au sein dans leurs vies. La reconstruction de sein est partie ...

Die menschliche Haut in der modernen Kunst
Dr. Ralph Ubl, Professor für neuere Kunstgeschichte an der Universität Basel, stellte sich der schwierigen Herausforderung, einem Raum voller erwartungsvoller Dermatologen das Organ Haut ...