Genetische Aspekte im Management von uroonkologischen Patienten
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
In den letzten Jahren haben sich mit der Entzifferung des menschlichen Genoms sowie der Etablierung von Next-Generation- Sequencing-Technologien die Möglichkeiten zur Diagnostik von genetischen Veränderungen in der Medizin rasant entwickelt. Mittlerweile wurden zahlreiche erbliche Tumorerkrankungen beschrieben. Deren Diagnostik und klinisches Management betreffen auch uroonkologische Patienten.
Keypoints
-
Prostatakarzinome, Nierenzellkarzinome und Urothelkarzinome können zu einem beträchtlichen Anteil auf Keimbahnmutationen in Tumorsuppressorgenen zurückgeführt werden.
-
Das Ergebnis der molekulargenetischen Untersuchung der Erkrankten beeinflusst die weitere Therapie (Operationen, Checkpoint-Inhibitoren, PARP-Inhibitoren).
-
Eine genetische Beratung und Abklärung ermöglichen auch gesunden Mutationsträgern die Durchführung von Untersuchungen zur Früherkennung und gegebenenfalls eine frühzeitige Behandlung.
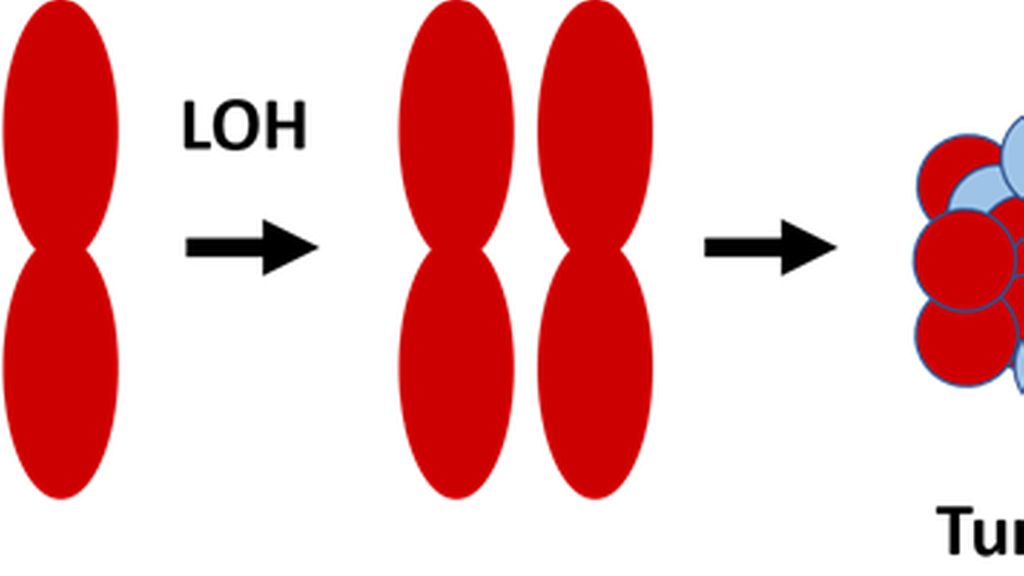
Einem Tumorprädispositionssyndrom, also einer erblichen Tumorerkrankung, liegt in den meisten Fällen eine Mutation in einem Tumorsuppressorgen zugrunde. Nach der 2-Hit-Hypothese von Knudson bedarf es der Mutation beider Allele eines Tumorsuppressorgenes zur Entartung einer Zelle. Liegt bereits seit Geburt ein verändertes Allel vor, wie das bei Keimbahnmutationen der Fall ist, ist die Wahrscheinlichkeit für den zweiten „Hit“ im Laufe des Lebens höher. In diesem Fall treten Krebserkrankungen häufiger und früher in der Lebensspanne auf (Abb. 1).1
Die genetische Abklärung von erblichen Tumorprädispositionssyndromen nimmt in der Urologie einen immer größeren Stellenwert ein. Zum einen kann das Wissen der Familienangehörigen um ihre erhöhte Erkrankungsneigung dazu beitragen, frühzeitige Screening-Untersuchungen in Anspruch zu nehmen. Zum anderen können erbliche Krebserkrankungen sowohl die Therapiewahl als auch deren Sequenzierung beeinflussen. Die immer präzisere Abklärung von Patienten mit onkologischen Erkrankungen ermöglicht eine zielgerichtetere und personalisierte Therapie. Eine Tatsache, die den betreuenden Uroonkologen eine sehr hohe Expertise abverlangt und für die betroffenen Patienten neue Hoffnung bringt.

Abb. 1: Knudson-2-Hit-Hypothese: Für die Entstehung von Krebs sind Mutationen an beiden Allelen erforderlich. Die Schädigung des zweiten Allels führt zum Verlust der Heterozygotie (LOH). Patienten mit erblichen Mutationen in der Keimbahn entwickeln frühzeitiger Tumoren, da die Schädigung eines Allels bereits von Geburt an in allen Körperzellen vorliegt1
Prostatakarzinom
In der Entstehung von Prostatakrebs spielt neben Alter auch die familiäre Veranlagung bei bis zu 20% aller Patienten eine Rolle.2 Bei etwa 10% der Prostatakarzinompatienten liegt eine monogenetisch vererbte Mutation zugrunde. Die betroffenen Gene sind zum größten Teil in der DNA-Reparatur involviert,3–6 wie z.B. BRCA2, ATM, BRCA1, CHEK2, NBN, PALB2, RAD51, FANCA, ATR oder BRIP. In einer wegweisenden Publikation von Pritchard et. al wurde in 44% der erblich bedingten Prostatakarzinome eine Keimbahnmutation im BRCA2-Gen nachgewiesen. Die BRCA-Gene bzw. ATM sind bereits bei familiärem Brust- und Eierstockkrebs bekannt und außerdem mit einem höheren Risiko für die Entstehung von Pankreaskarzinomen, männlichem Brustkrebs sowie Prostatakarzinomen assoziiert. Auch die mit dem Lynch-Syndrom assoziierten Gene der DNA-Mismatch-Reparatur (MSH2, MSH6) dürften in geringerem Ausmaß auch die Entstehung von Prostatakrebs begünstigen.6
Sowohl Mutationen in der homologen Rekombination als auch DNA-Mismatch-Reparatur eröffnen Möglichkeiten für zielgerichtete systemische Therapien. Checkpoint-Inhibitoren oder PARP-Inhibitoren zeigten sich sowohl bei somatischen als auch die Keimbahn betreffenden Mutationen als vielversprechend. Erst kürzlich konnte ein verlängertes progressionsfreies Überleben von Patienten mit Mutationen in DNA-Reparaturgenen durch den PARP-Inhibitor Olaparib nachgewiesen werden. Olaparib wurde hierbei in der zweiten Linie mit Abirateron oder Enzalutamid bei Patienten mit metastasiertem kastrationsresistentem Prostatakarzinom verglichen.7
HOXB13 und Einzelnukleotid-Polymorphismen
Durch Mutationen der DNA-Reparaturgene können nicht alle familiären Prostatakarzinome erklärt werden. So ist z.B. bei Patienten mit Mutationen im HOXB13-Gen ebenfalls ein erhöhtes Erkrankungsrisiko festgestellt worden. Die Mutation betrifft nur einen geringen Anteil der Patienten mit Prostatakarzinom (0,73%), ist jedoch mit einer beträchtlichen Penetranz von etwa 35% vergesellschaftet und führt zu einer früheren Manifestation der Erkrankung.8 Des Weiteren dürften SNP unter anderem im Androgenrezeptor oder in Onkogenen wie MYC zu den polygenen Ursachen des familiären Prostatakarzinoms zählen. Hierbei bedarf es weiterer genomweiter Assoziationsstudien, um das genaue Zusammenspiel der betroffenen Gene zu erforschen .9
Urothelkarzinom des oberen Harntraktes
Das Urothelkarzinom des oberen Harntraktes (UTUC) ist zu einem nicht unerheblichen Prozentsatz auf eine Keimbahnmutation aus dem Lynch-Syndrom-Spektrum zurückzuführen.10 Beim Lynch-Syndrom handelt es sich um eine autosomal dominante Vererbung von Mutationen der DNA-Mismatch-Reparaturgene. Da Fehler in der DNA nicht mehr ausgebessert werden, kommt es zur malignen Entartung und somit zur Tumorentstehung. Mutationsträger haben ein Lebenszeitrisiko von bis zu 80%, eine mit dem Lynch-Syndrom assoziierte Krebserkrankung (wie z.B. kolorektale Karzinome, Endometriumkarzinom, Ovarialkarzinom, Pankreaskarzinom und UTUC) diagnostiziert zu bekommen.11
Durch das hohe Erkrankungsrisiko ist daher bei UTUC-Patienten eine gezielte Anamneseerhebung wichtig. In weiterer Folge können betroffene Patienten und ihre Familienangehörigen von einer genetischen Abklärung und den darauffolgenden Krebsfrüherkennungsuntersuchungen profitieren. Da Checkpoint-Inhibitoren vor allem bei einer hohen Mikrosatelliteninstabilität ein gutes Ansprechen zeigten, können Patienten mit UTUC aufgrund eines Lynch-Syndroms zielgerichtete Therapien wie Pembrolizumab erhalten.12
Urothelkarzinom der Harnblase
Das Urothelkarzinom der Blase kann nicht auf ein klassisches erbliches Tumorsyndrom zurückgeführt werden. Allerdings konnte gezeigt werden, dass erstgradig Verwandte von an einem Blasenkarzinom Erkrankten ein beinahe doppelt so hohes Risiko tragen, selbst an Blasenkrebs zu erkranken.13,14 Bisher konnten aus dem Lynch-Syndrom-Spektrum lediglich Mutationen im MSH2-Gen mit der Entstehung von Urothelkarzinomen der Blase in Zusammenhang gebracht werden.15
Einzelnukleotid-Polymorphismus
Die Entstehung von Urothelkarzinomen der Blase kann zu einem beträchtlichen Teil auf Umweltfaktoren zurückgeführt werden. Da jedoch der Anteil an Patienten mit Blasenkrebs zum Beispiel unter Rauchern relativ gering ist, geht man davon aus, dass polygenetische Veränderungen bei bestimmten Personengruppen zu einem erhöhten Auftreten von Urothelkarzinom führen. Genomweite Assoziationsstudien konnten Einzelnukleotid-Polymorphismen (SNP) nachweisen, die bei Blasenkarzinompatienten in der Nähe von Onkogenen oder Tumorsuppressorgenen liegen (z.B. MYC, TP63, FGFR3). Bisher geht man davon aus, dass viele dieser SNP eine Rolle in der Entgiftung von Karzinogenen, der Zellzykluskontrolle oder der Erhaltung der DNA-Integrität spielen. Des Weiteren können SNP auch als Enhancer der Transkription fungieren. Allerdings ist die Wahrscheinlichkeit, dass einzelne SNP für die Krebsentstehung verantwortlich sind, so gering, dass derzeit genetische Untersuchungen auf SNP nicht zielführend sind.16
Nierenzellkarzinom
Etwa 5 bis 8% aller Nierenzellkarzinome (RCC) lassen sich auf eine erbliche Mutation zurückführen.17 Hinweisgebend können neben einer positiven Familienanamnese ein frühes Erkrankungsalter oder das Auftreten von bilateralen oder multizentrischen Nierenzellkarzinomen sein. Zusätzlich können anamnestisch weitere Tumorerkrankungen oder spezielle Organmanifestationen wie etwa Hämangioblastome der Retina sowie histopathologische oder immunhistochemische Eigenschaften des Tumors wie eine nichtklarzellige Histologie des RCCs auf erbliche Syndrome hindeuten. Das mediane Erkrankungsalter für hereditäre Nierenzellkarzinome liegt bei 37 Jahren. Hereditäre Nierenzellkarzinome sind unter anderem mit dem Von-Hippel-Lindau-Syndrom (VHL), dem hereditären papillären RCC, dem Birt-Hogg-Dubé-Syndrom, der hereditären Leiomyomatose mit Nierenzellkarzinom sowie dem Succinat-Dehydrogenase-defizienten Nierenzellkarzinom assoziiert.
Von-Hippel-Lindau-Syndrom
Das erbliche VHL zeichnet sich durch ein erhöhtes Auftreten von Nierenzellkarzinomen als auch Phäochromozytomen sowie Angiomen der Retina und des Cerebellums aus.18 Kennzeichnend ist vor allem die hohe Penetranz – bis zu 70% der Träger einer Keimbahnmutation im VHL-Gen entwickeln im Laufe ihres Lebens ein Nierenzellkarzinom.19 Mit dem VHL-Syndrom assoziierte Nierenzellkarzinome unterscheiden sich von sporadischen Tumoren dadurch, dass sie bilateral bzw. multizentrisch in jungem Alter auftreten. Diese Charakteristik hat auch Einfluss auf die Therapieempfehlungen. Da Tumoren oft in einem wenig aggressiven Stadium diagnostiziert werden, im Laufe des Lebens aber mit weiteren Rezidiven zu rechnen ist, sollte eine nierenerhaltende Therapie im Vordergrund stehen.20,21 Die NCCN-Guidelines empfehlen eine aktive Überwachung bei Tumoren bis zur Größe von 3,0cm. Bei einer Tumorgröße über 3,0cm erhöht sich das Risiko für eine Metastasierung, weshalb hier eine chirurgische Intervention indiziert ist, die vorzugsweise nephronsparend zu planen ist (Tab. 1).22

Tab. 1: Übersicht über die erblichen Nierenzellkarzinome. Zusammengefasst aus Schmidt LS et al.18, Costa WH et al.42, Maher ER43
Tuberöse-Sklerose-Komplex
Beim Tuberöse-Sklerose-Komplex (TSC) handelt es sich um eine autosomal dominante neurokutane Erkrankung. Bei den Betroffenen treten gutartige Hamartome auf, die sich in beinahe jedem Organ manifestieren können. Die Trias aus Epilepsie, mentaler Retardierung sowie Angiofibromen der Haut ist hinweisend für eine Diagnose, diese wird jedoch anhand von festgelegten Charakteristiken getroffen (z.B. Angiomyolipome, Angiofibrome des Gesichts).23 Neben Erkrankungen des zentralen Nervensystems treten beim TSC bei 40–80% der Patienten Angiomyolipome (AML) der Nieren auf. Dies sind gutartige Raumforderungen der Niere, die keiner aktiven Therapie bedürfen, sie können jedoch einbluten oder aufgrund ihrer Größe Symptome verursachen.24, 25 Neben AML können auch Nierenzysten bereits im Kindesalter auftreten. Aktive Überwachung ist die geeignetste Option bei den meisten Patienten mit AML. In einer Kohorte von Patienten unter aktiver Überwachung zeigten nur 11% ein Wachstum des AML, bei 2% kam es zu spontanen Blutungen. Eine aktive Therapie sollte im Falle von anhaltenden Schmerzen, rupturiertem AML (akute oder rezidivierende Blutungen) oder im Falle von sehr großen AML durchgeführt werden. Bei Patienten mit einem erhöhten Risiko für abdominelle Traumata, Frauen im gebärfähigen Alter und wenn eine Notfallversorgung nicht gewährleistet ist, sollte die chirurgische Sanierung diskutiert werden. Für eine systemische Therapie bzw. um das Wachstum der TSC-assoziierten Tumoren einzuschränken, stehen mTOR-Inhibitoren wie Sirolimus und Everolimus zur Verfügung.26, 27
Birt-Hogg-Dubé
Das Birt-Hogg-Dubé-Syndrom (BHD) wird autosomal dominant vererbt. Es findet sich eine Mutation im Follikulin-Gen. Charakteristika sind multiple kutane Fibrofollikulome, Lungenzysten, Auftreten eines Spontanpneumothorax, kolorektale Tumoren sowie unterschiedliche Formen des Nierenzellkarzinoms (chromophobes RCC, Onkozytome und hybrid-onkozytisch-chromophobe Mischtumoren).28–30 Die Penetranz beim BHD liegt für RCC lediglich bei etwa 25%, mit einem mittleren Ersterkrankungsalter von 50 Jahren.31, 32 Die meisten Nierentumoren bei BHD zeigen einen langsamen Verlauf, weswegen hier auch in erster Linie eine aktive Überwachung bei Tumoren von unter 3cm Größe und im Falle einer Resektion eine nierenerhaltende Therapie empfohlen werden.22, 33
Hereditäres papilläres Nierenzellkarzinom
Eine äußerst seltene Form des erblichen Nierenzellkarzinoms stellt das hereditäre papilläre Nierenzellkarzinom (HPRCC) dar. Hier ist eine autosomal dominant vererbte Missense-Mutation des MET-proto-Onkogens krankheitsverursachend,34 und die Nierenzellkarzinome treten im Alter von etwa 45 Jahren auf. Bei diesem erblichen Tumorprädispositionsyndrom sind abgesehen von der Niere keine weiteren Organsysteme betroffen. Aufgrund einer rasch fortschreitenden Niereninsuffizienz liegt die mittlere Lebenserwartung bei Mutationsträgern bei lediglich 52 Jahren.35 Bei metastasiertem Nierenzellkarzinom zeigen neue Studienergebnisse vielversprechende Resultate mit MET-Inhibitoren.36,37
Hereditäre Leiomyomatose mit Nierenzellkarzinom und Succinat-Dehydrogenase-defizientes Nierenzellkarzinom
Liegt eine hereditäre Leiomyomatose mit Nierenzellkarzinom (HLRCC) vor, weisen Patienten neben Nierenzelltumoren auch zahlreiche kutane als auch uterine Leiomyome auf. Weiters ist die ursächliche Mutation der Fumarathydratase auch verantwortlich für die Entstehung seltener erblicher Phäochromozytome.38 Das Risiko, ein Nierenzellkarzinom zu entwickeln, liegt bei Mutationsträgern bei ca. 15%. Es handelt sich in diesem Fall meist um aggressive Tumoren, welche frühzeitig metastasieren.22,39 Ähnlich verhält es sich beim Succinat-Dehydrogenase-defizienten Nierenzellkarzinom (SDH-RCC), welches durch ein frühzeitiges Auftreten von aggressiven Nierenzelltumoren, Phäochromozytomen und Paragangliomen im HNO-Bereich gekennzeichnet ist.40
Cowden-Syndrom
Mutationen im PTEN-Gen sind für das Cowden-Syndrom verantwortlich. Bei diesem Tumorprädispositionssyndrom haben Mutationsträger ein erhöhtes Risiko, an Brust-, Nierenzell- und Schilddrüsenkrebs zu erkranken. Da knapp 90% der Mutationsträger im Laufe ihres Lebens eine Tumorerkrankung diagnostiziert bekommen, werden beim Cowden-Syndrom frühzeitige Screening-Untersuchungen empfohlen (Nierenultraschall ab 40 Jahren, Brust-MRT ab 30 Jahren, Schilddrüsenultraschall ab 18 Jahren).41
Fazit
Die Uroonkologie wird sich in Zukunft immer mehr mit erblichen Krebserkrankungen beschäftigen müssen. Durch Next-Generation Sequencing können krankheitsverursachende Gene idenfiziert und diagnostiziert werden. Dies eröffnet Klinikern die Möglichkeit, Patienten und ihre Angehörigen in Bezug auf erbliche Krebserkrankungen abzuklären, und erfordert in weiterer Folge die langfristige Betreuung der Mutationsträger durch entsprechende Früherkennungsuntersuchungen. Die Möglichkeiten der Abklärung stoßen in Bezug auf die klinische Konsequenz manchmal auf ethische Grenzen. Erbliche Syndrome mit inkompletter Penetranz können für gesunde Mutationsträger eine psychische Belastung darstellen. Aus diesem Grund spielt bei einer genetischen Abklärung das empathische ärztliche Gespräch eine wichtige Rolle. Während der genetischen Beratung und Aufklärung können Patienten festlegen, ob sie Befunde ohne klinische Konsequenz mitgeteilt bekommen möchten, oder Angehörige angeben, welchen das Ergebnis übermittelt werden darf. Mit der Entwicklung zielgerichteter Therapien wie PARP-Inhibitoren oder Checkpoint-Inhibitoren hat die genetische Abklärung auch Einfluss auf Therapieoptionen. Auch die Entscheidung für eine operativen Tumorexzision kann durch eine molekulargenetische Untersuchung beeinflusst werden. Somit bietet die genetische Abklärung dem Uroonkologen eine zusätzliche Möglichkeit, patienten-orientierte Diagnostik durchzuführen und entsprechende Therapieentscheidungen zu treffen.
Literatur:
1 Knudson AG Jr.: Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971; 68(4): 820-3 2 Hemminki K et al.: How common is familial cancer? Ann Oncol 2008; 19(1): 163-7 3 Castro E et al.: Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol 2013; 31(14): 1748-57 4 Castro E et al.: Effect of BRCA mutations on metastatic relapse and cause-specific survival after radical treatment for localised prostate cancer. Eur Urol 2015; 68(2): 186-93 5 Mitra A et al.: Prostate cancer in male BRCA1 and BRCA2 mutation carriers has a more aggressive phenotype. Br J Cancer 2008; 98(2): 502-7 6 Pritchard CC et al.: Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016; 375(5): 443-53 7 de Bono J et al.: Olaparib for metastatic castration-resistant prostate cancer. Reply. N Engl J Med 2020; 383(9): 891 8 Handorf E et al.: Prevalence of the HOXB13 G84E mutation among unaffected men with a family history of prostate cancer. J Genet Couns 2014; 23(3): 371-6 9 Heidegger I et al.: Hereditary prostate cancer - Primetime for genetic testing? Cancer Treat Rev 2019; 81: 101927 10 Carlo MI et al.: Cancer susceptibility mutations in patients with urothelial malignancies. J Clin Oncol 2020; 38(5): 406-14 11 Stoffel E et al.: Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology 2009; 137(5): 1621-7 12 Huyghe N et al.: Immunotherapy with immune checkpoint inhibitors in colorectal cancer: what is the future beyond deficient mismatch-repair tumours? Gastroenterol Rep (Oxf) 2020; 8(1): 11-24 13 Kramer AA et al.: Familial aggregation of bladder cancer stratified by smoking status. Epidemiology 1991; 2(2): 145-8 14 Aben KK et al.: Familial aggregation of urothelial cell carcinoma. Int J Cancer 2002; 98(2): 274-8 15 van der Post RS et al.: Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet 2010; 47(7): 464-70 16 Golka K et al.: Genetic variants in urinary bladder cancer: collective power of the „wimp SNPs“. Arch Toxicol 2011; 85(6): 539-54 17 Menko FH et al.: Diagnosis and management of hereditary renal cell cancer. Recent results. Cancer Res 2016; 205: 85-104 18 Schmidt LS et al.: Genetic predisposition to kidney cancer. Semin Oncol 2016; 43(5): 566-74 19 Nordstrom-OʼBrien M et al.: Genetic analysis of von Hippel-Lindau disease. Hum Mutat 2010; 31(5): 521-37 20 Linehan WM et al.: The metabolic basis of kidney cancer. Semin Cancer Biol 2013; 23(1): 46-55 21 Metwalli AR et al.: Nephron-sparing surgery for multifocal and hereditary renal tumors. Curr Opin Urol 2014; 24(5): 466-73 22 Shuch B et al.: The surgical approach to multifocal renal cancers: hereditary syndromes, ipsilateral multifocality, and bilateral tumors. Urol Clin North Am 2012; 39(2): 133-48 23 Roach ES et al.: Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998; 13(12): 624-8 24 Chonko AM et al.: Renal involvement in tuberous sclerosis. Am J Med 1974; 56(1): 124-32 25 Garrick R et al.: Demyelination of the brain in tuberous sclerosis: computed tomography evidence. Mayo Clin Proc 1979; 54(10): 685-9 26 Kohrman MH: Emerging treatments in the management of tuberous sclerosis complex. Pediatr Neurol 2012; 46(5): 267-75 27 Bissler JJ et al.: Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2013; 381(9869): 817-24 28 Nickerson ML et al.: Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell 2002; 2(2): 157-64 29 Menko FH et al.: Birt-Hogg-Dube syndrome: diagnosis and management. Lancet Oncol 2009; 10(12): 1199-206 30 Boris RS et al.: The impact of germline BHD mutation on histological concordance and clinical treatment of patients with bilateral renal masses and known unilateral oncocytoma. J Urol 2011; 185(6): 2050-5 31 Dal Sasso AA et al.: Birt-Hogg-Dube syndrome. State-of-the-art review with emphasis on pulmonary involvement. Respir Med 2015; 109(3): 289-96 32 Toro JR et al.: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J Med Genet 2008; 45(6): 321-31 33 Stamatakis L et al.: Diagnosis and management of BHD-associated kidney cancer. Fam Cancer 2013; 12(3): 397-402 34 Schmidt L et al.: Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet 1997; 16(1): 68-73 35 Zbar B et al.: Hereditary papillary renal cell carcinoma: clinical studies in 10 families. J Urol 1995; 153(3 Pt 2): 907-12 36 Choueiri TK et al.: Biomarker-based phase II trial of savolitinib in patients with advanced papillary renal cell cancer. J Clin Oncol 2017; 35(26): 2993-3001 37 Choueiri TK et al.: Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol 2013; 31(2): 181-6 38 Clark GR et al.: Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab 2014; 99(10): E2046-50 39 Linehan WM, Rouault TA: Molecular pathways: Fumarate hydratase-deficient kidney cancer--targeting the Warburg effect in cancer. Clin Cancer Res 2013; 19(13): 3345-52 40 Ricketts C et al.: Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 2008; 100(17): 1260-2 41 Riegert-Johnson DL et al.: Cancer and Lhermitte-Duclos disease are common in Cowden syndrome patients. Hered Cancer Clin Pract 2010; 8(1): 6 42 Costa WH et al.: Urological cancer related to familial syndromes. Int Braz J Urol 2017; 43(2): 192-201 43 Maher ER: Hereditary renal cell carcinoma syndromes: diagnosis, surveillance and management. World J Urol 2018; 36(12): 1891-8
Das könnte Sie auch interessieren:

Aktuelle Entwicklungen und Erkenntnisse beim Urothelkarzinom
Auf dem diesjährigen Genitourinary Cancers Symposium der American Society of Clinical Oncology (ASCO-GU-Kongress) wurden bedeutende Fortschritte in der Diagnose und Behandlung des ...

Aktuelles aus der 7. Version der S3-Leitlinie: wesentliche Leitlinienänderungen
Im Mai 2024 wurde die Prostatakarzinom-S3-Leitlinie unter der Federführung der Deutschen Gesellschaft für Urologie e.V. (DGU) im Rahmen des Leitlinienprogramms Onkologie in ihrer 7. ...

Neues vom ASCO GU zum Prostatakarzinom
Im Rahmen des ASCO GU 2025 in San Francisco wurden eine Vielfalt von neuen praxisrelevanten Studien zum Prostatakarzinom präsentiert. Mit Spannung wurde unter andem auch auf die finalen ...