
Neurofibromatose Typ 1 im Kindesalter
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Menschen mit Neurofibromatose Typ 1 sind nicht per se krank. Betroffene weisen aber ein erhöhtes Risiko für Tumoren auf wie auch für nichtonkologische Manifestationen.
Neurofibromatose Typ 1 (NF1) ist ein genetisches Tumorprädispositionssyndrom, das zu den Phakomatosen bzw. neurokutanen Syndromen gezählt wird. Mit einer Inzidenz von etwa 1:2500 zählt die NF1 zu den häufigsten seltenen Erkrankungen (per definitionem <1:2000).1 Zugrunde liegt eine pathologische Veränderung (Mutation oder Deletion) im NF1-Gen. Die Erkrankung wird autosomal dominant vererbt, bei etwa 50% der Betroffenen handelt es sich jedoch um de-novoMutationen, d.h.,keiner der Eltern ist betroffen. Das NF1-Gen kodiert für das Tumorsuppressor-Molekül Neurofibromin. Dieses greift bremsend in den MAP-Kinase-Signalweg ein, indem es Ras deaktiviert. Bei einem Defekt beider NF1-Allele (d.h. einer somatischen Inaktivierung des zweiten gesunden Allels, „Two-Hit-Hypothese“) kommt es zu einer Überaktivierung des MAP-Kinase-Pathways. Die Aktivierung der Downstream-Signalmoleküle MEK und ERK führt schließlich u.a. zu einer vermehrten Proliferation der Zelle. Dies führt zur Entstehung von Tumoren, aber auch mannigfaltigen anderen, nichtonkologischen Symptomen, die in diesem Artikel aufgeführt werden.
Zu betonen ist, dass die NF1 nichts mit der sogenannten NF2-assoziierten Schwannomatose (früher Neurofibromatose Typ 2 oder NF2) zu tun hat und es sich um zwei genetisch wie klinisch völlig distinkte Erkrankungen handelt!
Diagnosestellung
Bei der NF1 können viele unterschiedliche Symptome auftreten. Die wichtigsten flossen in die Diagnosekriterien ein. Eine Genotyp-Phänotyp-Korrelation ist mit wenigen Ausnahmen bis dato nicht möglich. Sicher ist, dass Mikrodeletionen des NF1-Gens und u.U. auch flankierender Gene (z.B. SUZ12) zu einem schweren Verlauf prädisponieren.2
Die mehr als 30 Jahre alten Diagnosekriterien der NF1 („NIH-Kriterien“) wurden in einem internationalen Konsensus-Prozess überarbeitet und 2021 von Legius et al. publiziert (siehe Tab. 1).3
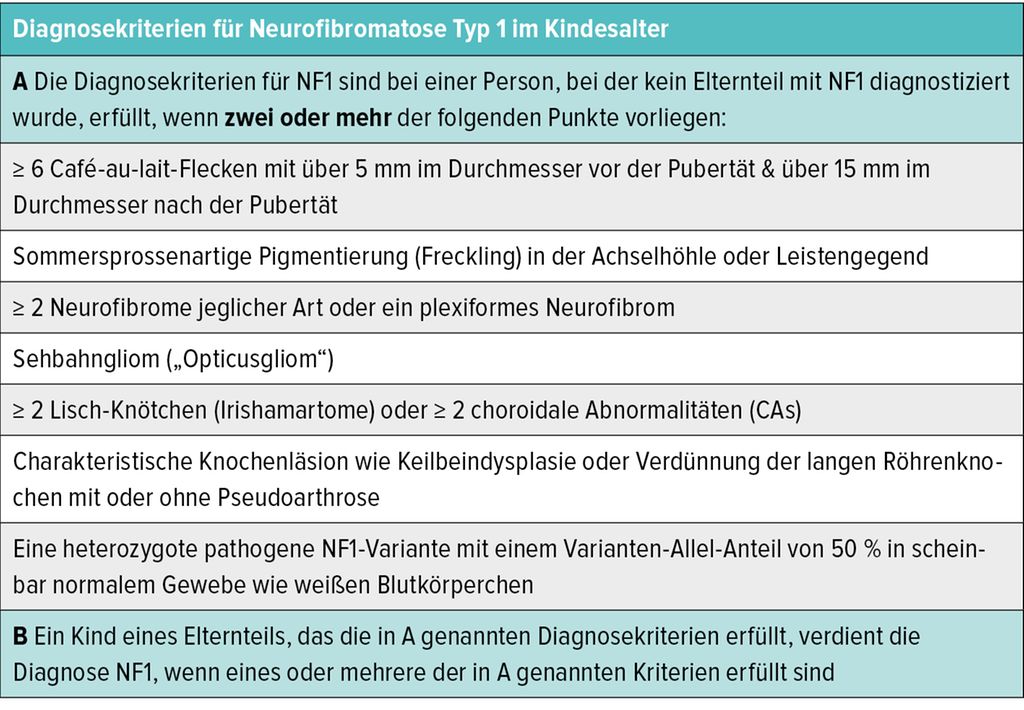
Tab. 1: Diagnosekriterien (mod. nach Legius et al.)3
Das häufigste Symptom stellen hyperpigmentierte, meist scharf begrenzte Maculae dar, sogenannte Café-au-lait-Flecken (CALM). Diese treten meist im 1. Lebensjahr auf und können lebenslang an Zahl und Größe zunehmen. Sie entarten nicht und sind so gut wie bei allen Betroffenen vorhanden. Vereinzelte sporadische CALM sind häufig in der gesunden Population, Anzahl und Konfiguration sind jedoch oft wegweisend, um den Verdacht auf das Vorliegen einer NF1 zu stellen.4 Die Liste an möglichen Differenzialdiagnosen für CALM ist lang, NF1 ist jedoch die bei Weitem häufigste Ursache syndromaler CALM.5 Als wichtige Differenzialdiagnosen sind zu nennen: Das Legius-Syndrom(SPRED1-Mutation)ist mit der NF1 verwandt und führt zu CALM und Lernschwierigkeiten, u.U. Lipomen, aber keinen Tumoren. Beim McCune-Albright-Syndrom (GNAS-Mutation, meist im Mosaik) liegt die Trias aus multiplen ossären fibrösen Dysplasien, Café-au-lait-Flecken (meist mit der Mittellinie begrenzt) und Pubertas praecox (und u.U. weiteren Hormonstörungen) vor. Beim konstitutionellen Mismatch-Repair-Gendefekt (CMMRD, biallelische Mutation im MSH1-, MSH2-, PMS2- oder MLH1-Gen) kommt es neben wenigen CALM frühzeitig zum Auftreten maligner Tumoren wie hochgradigen Gliomen, Medulloblastomen, Kolonpolypen und Kolonkarzinomen oder Lymphomen.
Freckling nennt man das meist im (Vor-)Schulalter auftretende sommersprossenartige Pigmentmuster axillär oder inguinal. Für die Diagnosestellung müssen laut den neuen Richtlinien CALM bzw. Freckling bilateral auftreten (aufgrund der Möglichkeit von NF1-Mutationen im Mosaik).
Sehbahngliome (OPG, „optic pathway glioma“) inkl. Opticusgliomen kommen häufig (15–20%) bei Kindern mit NF1 vor,mit einem Prädilektionsalter zwischen 2 und 8 Jahren. Während OPG in etwa der Hälfte der Fälle asymptomatisch bleiben, so können sie auch bis zur Erblindung führen und müssen daher frühzeitig erkannt und bei Symptomatik behandelt werden.
Typische Knochenveränderungen wie anterolaterales Bowing der Tibia („Tibiadysplasie“) oder anderer langer Röhrenknochen betreffen ca. 5% aller Kinder. Sphenoiddysplasien mit möglicherweise auftretenden Gesichtsasymmetrien sind seltener. Diese Veränderungen sind entweder bei Geburt bereits da (z.B. Keilbeindysplasie) oder manifestieren sich in den ersten 1,5 Lebensjahren, insbesondere wenn die Kinder beginnen zu gehen und Gewicht auf die betroffenen Knochen zu verlagern.
Lisch-Knötchen sind gutartige Irishamartome, die pathognomonisch sind, aber ohne Krankheitswert. Sie treten erst im Schulalter auf. Choroidale Abnormalitäten (CA) sind häufig bei Erwachsenen mit NF1 und können mittels optischer Kohärenztomografie (OCT) oder Near-Infrared-Spektroskopie nachgewiesen werden. Eine pathophysiologische Bedeutung ist bis dato nicht bekannt. CA wurden neu in die Diagnosekriterien aufgenommen.
Die namensgebenden Neurofibrome (NF) sind gutartige Tumoren der Nerven, die fast alle Menschen mit NF1 betreffen. Kutane NF treten erst ab der Pubertät auf und nehmen stetig an Zahl und Größe zu. Sie können jucken und mechanisch oder ästhetisch störend sein. Plexiforme NF (PNF) können bei der Geburt vorhanden sein oder in jedem Lebensalter auftreten und kommenbei etwa 30–50% der Betroffenen vor.2 Sie können Dysästhesien auslösen, mechanisch störend sein (z.B. Atemwege, Abdomen, Extremitäten) oder ästhetisch entstellend. Sie können v.a. bei kleinen Kindern eine starke Wachstumstendenz zeigen, ihr Wachstumsverhalten kann jedoch nicht vorhergesagt werden. Von prognostischer Bedeutung ist das Risiko für maligneEntartung von PNF zumalignen peripheren Nervenscheidentumoren (MPNST) (in ca. 10% der PNF), MPNST können jedoch auch ohne fassbare PNF auftreten („lifetime risk“ 15% bei NF1).6 Symptome der Entartung können schnelles Wachstum eines PNF oder eines soliden Knotens innerhalb des PNF sein, ebenso (anhaltende) Schmerzen oder neuaufgetretener Funktionsausfall (z.B. Parästhesien oder Lähmungen).2 MPNST zählen nicht zu den Diagnosekriterien der NF1.
In den Diagnosekriterien von 2021 kam auch das Vorliegen einer pathogenen NF1-Gen-Veränderung hinzu, welche in nicht veränderten Zellen (z.B. Leukozyten) mit 50%igerAllelfrequenz nachgewiesen sein muss. Ein negativer Test schließt jedoch die Diagnose nicht aus. Der Goldstandard sieht immer noch die cDNA-Sequenzierung vor, mit Detektionsraten von 85–90%, während mittels NGS nur etwa zwei Drittel der Veränderungen detektiert werden können.
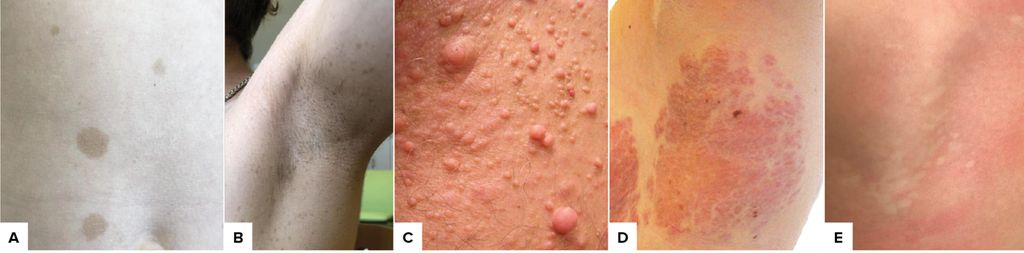
Abb. 1: A) Café-au-lait Flecken; B) axilläres Freckling; C) kutane Neurofibrome; D) plexiformes Neurofibrom; E) Naevus anaemicus
Weitere Symptome
Außer den in den Diagnosekriterien genannten Symptomen gibt es noch andere mögliche Manifestationen der NF1.7
Hirntumoren: Neben Sehbahngliomen können auch in anderen Regionen des ZNS niedriggradige Gliome (LGG) auftreten, die völlig asymptomatisch bleiben und daher manchmal nur beobachtet werden können, während sie bei Symptomatik oder Progredienz operativ oder medikamentös behandelt werden müssen. Mit zunehmendem Alter werden auch hochgradige Gliome (HGG) häufiger, die eine sehr ungünstige Prognose aufweisen.
Skoliosen betreffen ca. 10% aller Kinder und Jugendlichen mit NF1 und können in manchen Fällen rapid progredient sein und eine operative Versorgung notwendig machen.
Bluthochdruck ist relativ häufig bei Menschen mit NF1, auch im Kindesalter. Während die arterielle Hypertonie meist idiopathisch auftritt, so kann sie gehäuft mit Fibrodysplasien der Nierenarterien und konsekutiver Nierenarterienstenose einhergehen oder mit Phäochromozytomen, die bis zu 10% der Erwachsenen mit NF1 betrifft. Fibrodysplasien der zerebralen Arterien können in ca. 2% zu Moyamoya-ähnlichen zerebralen Gefäßerkrankungen und konsekutiven Minderdurchblutungen führen.
Neurokognitive Defizite (Teilleistungsstörungen sowie Konzentrations- und Aufmerksamkeitsstörungen [ADHD]) treten bei etwa der Hälfte der Patient:innen auf, auch Autismusspektrumstörungen (ASD) sind gehäuft.7Manche Kinder weisen einen sonderpädadogischen Förderbedarf auf, während andere Menschen mit NF1 ohne Schwierigkeiten ein Studium absolvieren können.
Auf der Haut können neben CALM und NF auch andere Manifestationen auftreten: Naevus anaemicus,Naevus spilus und Glomustumorender Finger.
Weiters sind auch andere Tumoren mit NF1 assoziiert, insgesamt liegt das Risiko für eine onkologische Erkrankung bei 60% über das gesamte Leben, wobei vor allem junge Erwachsene das höchste Risiko aufweisen.6 Frauen ab 30 weisen ein deutlich erhöhtes Brustkrebsrisiko auf. Gastrointestinale Stromatumoren (GIST) sind häufig bei erwachsenen NF1-Patient:innen. Sie sind meist asymptomatisch, können jedoch zu Abdominalgien und selten zu lebensbedrohlichen Blutungen führen. MPNST wurden bereits erwähnt. Die juvenile myelomonozytäre Leukämie ist eine sehr seltene, aber schwerwiegende Komplikation. Das Auftreten von malignen Tumoren führt dazu, dass die Lebenserwartung bei der NF1 um ca. 15 Jahre reduziert ist.
Management bei NF1 im Kindesalter
Das Wichtigste im Management von Menschen mit NF1 sind die Multi- und Interdisziplinarität und ein Case-Management. Während dieses in der Pädiatrie fast immer durch die Kinderärztin oder den Kinderarzt erfolgt (sei es durch pädiatrische Onkologen und Okologinnen bzw. Neuropädiater:innen), so muss im adulten Bereich oft erst ein entsprechendes Setting geschaffen werden. Es sind viele Disziplinen involviert: Pädiatrie, Ophthalmologie, plastische Chirurgie, Kinderchirurgie, Neurochirurgie, Neurologie, Onkologie, Orthopädie, Dermatologie, Radiologie, Nuklearmedizin, Genetik und evtl. auch noch spezifische Organfächer. Die Aufgabe des Case-Managers ist es, die Patient:innen und deren Familien zu begleiten und, falls indiziert, zum richtigen Zeitpunkt an die richtige Stelle zu lenken. Komplexe Fragestellungen müssen in interdisziplinären Boards (z.B. einem Board für periphere Nerventumoren oder einem Board für kindliche ZNS-Tumoren) besprochen und das Prozedere muss schließlich gemeinsam mit den Patient:innen und Familien festgelegt werden.
Ebenso wichtig wie die medizinische ist auch die psychologische, insbesondere auch neuropsychologische, und sozialarbeiterische Betreuung der Betroffenen und ihrer Familien. Eine weitere Ressource stellen Patient:innen-Organisationen dar, in Österreich der Verein NF-Kinder (www.nfkinder.at). Der Verein bietet Informationsangebote (z.B. Patiententagungen, Infobroschüren), aber auch gemeinsame Aktivitäten (z.B. Reitwochenenden, Trainingswochen) und Förderungen (z.B. soziales Kompetenztraining).
Im Jahr 2023 veröffentlichte eine Expertengruppe des Europäischen Referenznetzwerks für genetische Tumor-Prädispositions-Syndrome (ERN GENTURIS) eine Empfehlung für das Management bzgl. Vorsorge und Behandlung von NF1-assoziierten Tumoren.2 Deren Abhandlung würde den Rahmen dieses Artikels sprengen, weshalb auf die Publikation von Carton et al. verwiesen sei.2
In Österreich einigte sich eine Expertengruppe auf ein einheitliches Vorsorgemanagement für Kinder und Jugendliche, insbesondere auch bzgl. der nichttumoralen Manifestationen (A. Azizi et al., Manuskript in Vorbereitung). Am wichtigsten ist ein altersadaptiertes Management, da (wie oben ausgeführt) manche Komplikationen ein gewisses Prädilektionsalter aufweisen.
Bei jedem Besuch müssen auxiologische Daten, Blutdruck (s.o.) und Pubertätsstatus (Risiko für Pubertas praecox) erhoben werden. Es wird eine eingehende Anamnese erfragt (Entwicklungsstand, Sehentwicklung, Ausbildung und Auftreten von schnell wachsenden/schmerzenden Knoten [=Warnzeichen für bösartige Nerventumoren], Zeichen für ZNS-Tumoren,…),ein neurologischer Status sowie Hautstatuserhoben und aufeine mögliche Skoliose geachtet.
Bei Diagnosestellung oder bei der Abklärung wird eine genetische Testung vorgeschlagen.
Ab Diagnosestellung erfolgen aufgrund des Risikos für Sehbahngliome (Prädilektionsalter 2–8 Jahre) regelmäßige Augenkontrollen durch Spezialisten mit Erfahrung in der Untersuchung von Kindern – alle 6 Monate vor und alle 12 Monate nach dem 8. Lebensjahr bis zum 18. Lebensjahr. Bei visueller Symptomatik (z.B. Sehverschlechterung, Schielen, Exophthalmus) muss die Augenuntersuchung vorgezogen und eine Bildgebung mittels MRT durchgeführt werden. Da die Augenuntersuchung bei Kindern und insbesondere bei NF1 (Aufmerksamkeitsdefizit!) schwierig sein kann, führen manche Zentren ein Screening mittels MRT durch.
Eine MRT-Untersuchung des Gehirns muss außerdem auch bei Symptomen durchgeführt werden, die auf das Vorliegen eines Hirntumors schließen lassen können. Zu diesen gehören z.B. Nüchtern-Erbrechen mit Kopfschmerzen [cave! Erhöhtes Risiko von Aquädukt-Stenosen und Hydrocephalus!], neurologische Symptome wie zerebrale Anfälle, Sensibilitätsstörungen, Lähmungen oder Koordinationsstörungen. Zu beachten gilt allerdings, dass Kinder mit NF1 oft von den Eltern als tollpatschig oder ungeschickt beschrieben werden und dies typisch für die Erkrankung ist – ganz ohne ZNS-Tumor!
Orthopädische Kontrollen sollten jährlich erfolgen (ab dem Gehbeginn).
Eine neuropsychologische Diagnostik sollte nach Möglichkeit im Vorschulalter und bei Wechsel in die Mittelschule erwogen werden, ist jedoch spätestens bei Lernschwierigkeiten oder Aufmerksamkeitsdefizit indiziert. Auch kann eine Begleitung bei der Wahl der Schule bzw. des angestrebten Berufes zielführend sein.
Mit der Transition in den Erwachsenenbereich soll eine Ganzkörperbildgebung mittels MRT erfolgen, um die Last an internen Neurofibromen zu erheben, da hiervon das Risiko für spätere maligne Entartungen abgeschätzt werden kann. Im Erwachsenenalter muss besonders auf das Auftreten von malignen Veränderungen geachtet werden (z.B. Brustkrebsscreening mittels Mammografie-MRT). Mit zunehmendem Alter wächst oft auch der psychische Druck aufgrund der individuell sehr unterschiedlich empfundenen ästhetischen Beeinträchtigung durch kutane Neurofibrome, die je nach Leidensdruck angegangen werden müssen (z.B. Lasertherapie, psychologische Begleitung).
Therapeutische Überlegungen
NF1 ist eine lebensbegleitende Veranlagung und die allermeisten Manifestation sind aus onkologischer Sicht benigne. Somit ist es wichtig, die Balance zwischen einem Zuviel an Therapie und einer zu spät erfolgten Behandlung zu finden. Sehbahngliome (OPG) können mittels Chemotherapie (v.a. Carboplatin/Vincristin) bzgl. Tumoransprechen und Progressionsfreiheit gut behandelt werden, funktionell (Sehen!) ist das Ansprechen leider oft weniger gut. Es gibt verschiedene Risikofaktoren und eine frühzeitige Behandlung verbessert das funktionelle Outcome.8 Andererseits wird nur die Hälfte aller Kinder mit einem OPG überhaupt je symptomatisch und nur etwa ein Drittel von ihnen muss behandelt werden. Seltene maligne Hirntumoren müssen mittels Strahlen- und Chemotherapie behandelt werden.
Kutane Neurofibrome können je nach Leidensdruck (ästhetisch [!] oder mechanisch störend) mittels Skalpell oder Laser entfernt werden.9 Therapieentscheidungen bzgl. plexiformer Neurofibrome müssen unbedingt durch interdisziplinäre Expertenteams getroffen werden (u.a. mit NF-Spezialisten, Nervenchirurgen, Onkologen). Oftmals verhalten sich PNF klinisch silent und können beobachtet werden. Sollten PNF jedoch Probleme machen (z.B. durch Beeinträchtigung von Organen, Entstellung) so ist die operative Entfernung oder Reduktion das Mittel der ersten Wahl. Aufgrund von Ausdehnung, Lokalisation und des Einwachsens in umliegendes Gewebe sind PNF jedoch nicht selten schwer chirurgisch zu behandeln. Für Kinder und Jugendliche ist seit Kurzem ein MEK-Inhibitor in genau dieser Indikation zugelassen (inoperable, symptomatische PNF), der die Symptomatik verbessern und die Größe der PNF reduzieren kann.10 Die Nebenwirkungen bedürfen jedoch eines guten Managements im Alltag.
Die bösartigen MPNST müssen frühzeitig entdeckt und dann durch ein erfahrenes Team behandelt werden. Die Resektion in sano nach frühzeitiger Detektion kann kurativ sein, alle anderen Modalitäten bei fortgeschrittenem Befall zeigen ein schlechtes Ansprechen, sind jedoch meist dennoch indiziert (lokale Strahlentherapie, medikamentöse Therapie). Orthopädische Probleme müssen von den jeweiligen Experten betreut werden. Die Behandlung von Tibiadysplasien kann orthopädisch sehr herausfordernd sein (Orthesenversorgung und evtl. operative Sanierung), insbesondere wenn es nach Frakturen zum Auftreten sogenannter Pseudoarthrosen kommt. Bei Skoliosen kommen Physiotherapie, evtl. Mieder oder ein operatives Vorgehen infrage.Bezüglich neuropsychologischer Defizite können und sollen Kinder und Jugendliche laufend unterstützt werden, um eine adäquate Schulausbildung zu gewährleisten.
Zusammenfassung
Menschen mit NF1 sind nicht per se krank. Sie haben aber eine Veranlagung, die mit dem hohen Risiko einhergeht, Manifestationen wie Tumoren oder nichtonkologische Komplikationen zu entwickeln. Die Schwere der Erkrankung kann nicht vorhergesagt werden, daher sind regelmäßige altersadaptierte Kontrolluntersuchungen nötig. Die Betreuung muss durch eine bzw. einen Case-Manager:in innerhalb eines multi- und interdisziplinären Teams erfolgen, gemäß bestehenden Guidelines und doch so individualisiert wie möglich.
Literatur:
1 Hirbe AC, Gutmann DH: Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol 2014; 13(8): 834-43 2 Carton C et al.: ERN GENTURIS tumour surveillance guidelines for individuals with neurofibromatosis type 1. EClinicalMedicine 2023; 56: 101818 3 Legius E et al.: Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med 2021; 23(8): 1506-13 4 Ben-Shachar S et al.: Predicting neurofibromatosis type 1 risk among children with isolated café-au-lait macules. J Am Acad Dermatol 2017; 76(6): 1077-83 5 Albaghdadi M et al.: Value of a café-au-lait macules screening clinic: Experience from The Hospital for Sick Children in Toronto. Pediatr Dermatol 2022; 39(2): 205-10 6 Uusitalo E et al.: Distinctive cancer associations in patients with neurofibromatosis type 1. J Clin Oncol 2016; 34(17): 1978-86 7 Bergqvist C et al.: Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis 2020; 15(1): 37 8 Azizi AA et al.: NF1 optic pathway glioma: analyzing risk factors for visual outcome and indications to treat. Neuro Oncol 2021; 23(1): 100-11 9 Ly I et al.: Target product profile for cutaneous neurofibromas: clinical trials to prevent, arrest, or regress cutaneous neurofibromas. J Invest Dermatol 2023; 143(8): 1388-96 10 Gross AM et al.: Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med 2020; 382(15): 1430-42
Das könnte Sie auch interessieren:

Long-Acting-Konzepte als Meilenstein in der HIV-Therapie
In den letzten vier Jahrzehnten erfuhr die HIV-Therapie eine enorme Entwicklung und die Optionen für Menschen mit HIV haben sich grundlegend verändert. Aktuell dominiert ein neues ...

Das Mikrobiomvon Wunden und was Probiotika für uns tun können
Unser Darm steht mit einer Vielzahl an Mikroorganismen über unterschiedlichste Funktionsachsen, wie jener zwischen Darm und Haut, mit dem gesamten Organismus in Verbindung. Das Mikrobiom ...

Sexuell übertragbare Infektionen bei Kindern und Jugendlichen
Seit 2019 berichten die World Health Organization (WHO) und die Centersfor Disease Control and Prevention (CDC; USA) regelmäßig über einen dramatischen Anstieg der sexuell übertragbaren ...