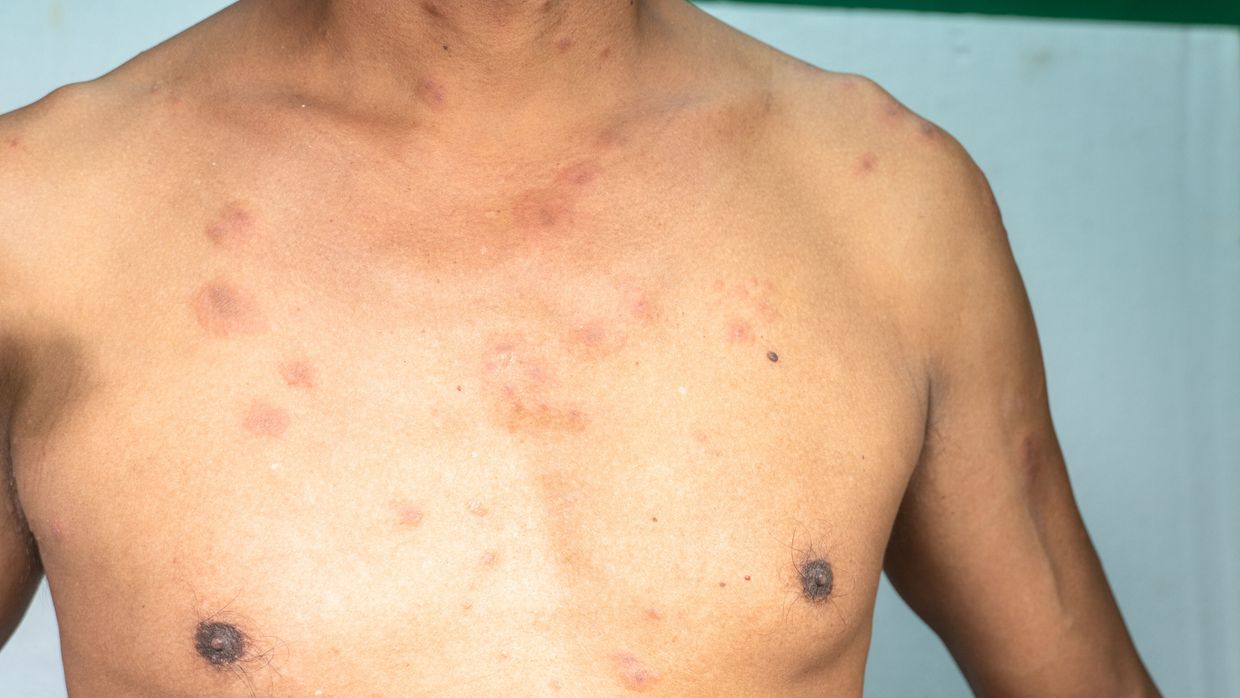
Néoplasme à cellules dendritiques plasmacytoïdes blastiques (BPDCN)
Le néoplasme à cellules dendritiques plasmacytoïdes blastiques (BPDCN) est une hémopathie maligne agressive qui est très rare, dont la fréquence représente environ 0,5% de l’ensemble des néoplasmes d’origine hématologique. Leslésions cutanées sont cependant les plus fréquentes, c’est pourquoi un diagnostic multidisciplinaire est essentiel.
Keypoints
-
Le BPDCN est une maladie rare. Le diagnostic doit être posé de manière multidisciplinaire par des dermatologues, des pathologistes et des hématologues.
-
Il est essentiel de penser au BPDCN en présence de lésions cutanées typiques et d’initier la procédure diagnostique correspondante.
-
En plus de la chimiothérapie conventionnelle, une nouvelle option thérapeutique est disponible avec le tagraxofusp pour permettre aux patient·es d’obtenir une rémission.
-
Encore aujourd’hui, seule une greffe de cellules souches permet d’envisager une survie à long terme.
Le BPDCN est dû à une prolifération clonale de précurseurs immatures de cellules dendritiques plasmacytoïdes. L’origine de ces cellules dendritiques plasmacytoïdes étant longtemps restée indéterminée, le nom de la maladie a évolué au fil du temps (p.ex. lymphome à cellules NK blastiques, leucémie à cellules NK CD4+ agranuleuses, leucémie/lymphome à cellules NK blastiques, tumeur hématodermique CD4+ CD56+ agranuleuse). Ce n’est qu’en 2016 que l’OMS a classé le BPDCN comme une pathologie à part entière parmi les tumeurs myéloïdes. Dans la classification actuelle de l’OMS, le BPDCN est également cité comme une pathologie à part entière parmi les tumeurs histiocytaires et à cellules dendritiques.
Épidémiologie
Le BPDCN survient principalement chez les personnes âgées, avec une incidence d’environ 0,04/100000. L’âge médian des patient·es est compris entre 61 et 71 ans. Le pourcentage de cas pédiatriques s’élève à 10 à 20%. On observe une prédominance chez les hommes de 3:1. Chez 20 à 30% des patient·es atteint·es de BPDCN, on trouve une tumeur myéloïde maligne antérieure ou coexistante, par exemple une leucémie aiguë myéloïde (LAM), une tumeur myélodysplasique ou une tumeur myélodysplasique/myéloproliférative. Le pronostic est défavorable, avec une survie moyenne d’environ neuf mois. Pour les patient·es qui ont répondu au traitement, la littérature indique une survie médiane de 12 à 15 mois. Le pronostic semble un peu plus favorable chez les enfants.
Tableau clinique et diagnostic
La plupart du temps, le BPDCN se manifeste par des lésions cutanées. Selon les séries de cas, la proportion de patient·es présentant une atteinte cutanée est estimée entre 70 et 100%. Des nodules érythémateux à livides uniques ou multiples, ou une éruption maculo-papuleuse sont des modifications typiques; des lésions de type hématome ont également été décrites. Le torse est souvent touché, mais la moitié des patient·es présentent des lésions maculo-papuleuses sur les extrémités et le visage. Les autres organes qui peuvent être atteints sont la moelle osseuse (60–90% des cas), le sang périphérique et les ganglions lymphatiques (40–50% des cas) (Tab. 1).
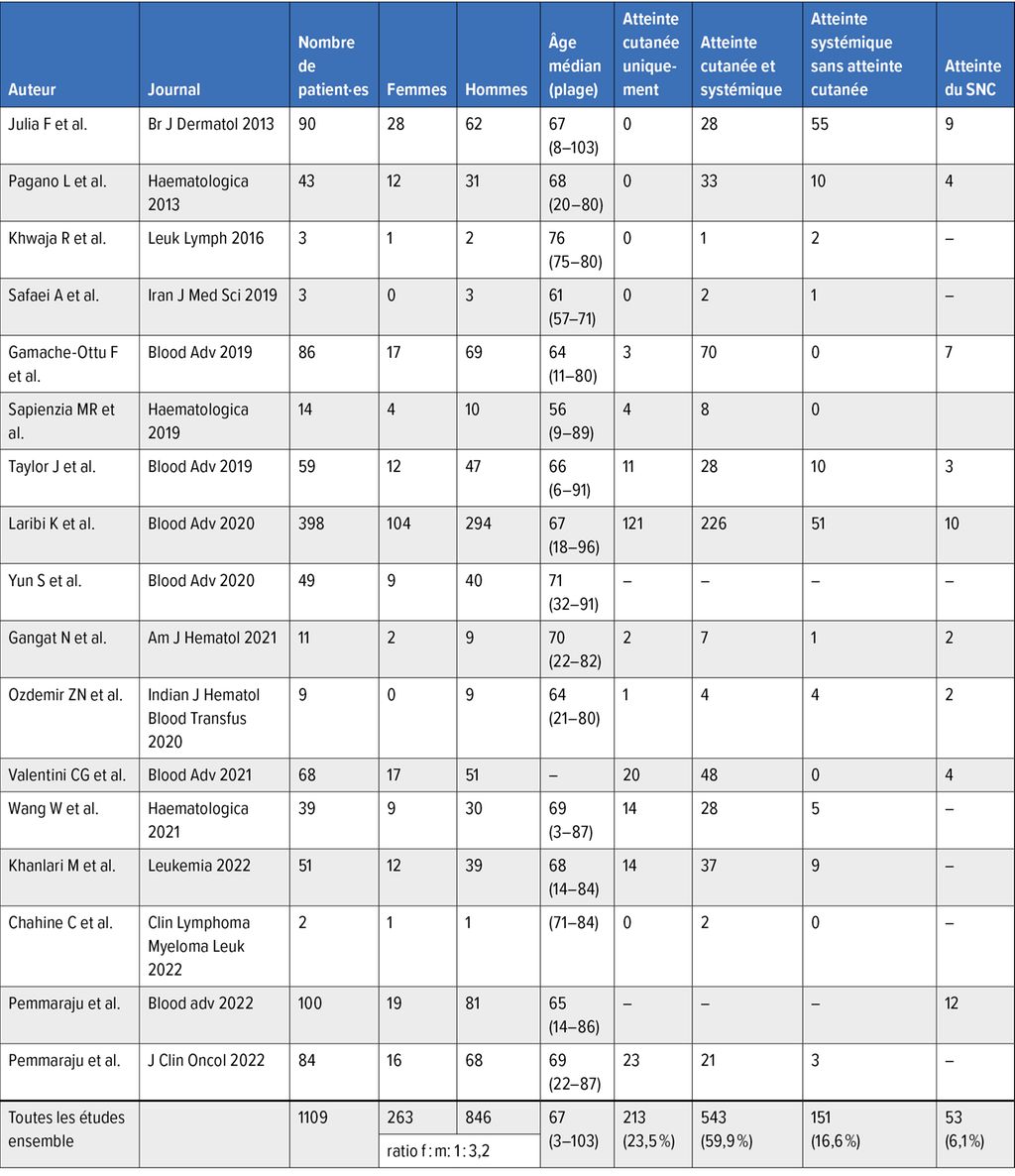
Tab. 1: Tableau clinique des patient·es dans différentes études
On suppose que la maladie se manifeste d’abord généralement au niveau de la peau (90% des patient·es) (Tab. 1). Une période variable de stabilité relative est suivie d’une propagation leucémique touchant de multiples organes, laquelle entraîne une détérioration rapide de l’état général et, souvent, le décès en peu de temps. Il est rare que la manifestation primaire du BPDCN soit extra-cutanée.
Le diagnostic est généralement posé grâce à l’analyse immunohistologique de la biopsie (peau, moelle osseuse, ganglions lymphatiques). Celle-ci révèle un infiltrat diffus et monomorphe de cellules blastiques de taille moyenne ainsi qu’un nombre élevé de mitoses (environ 80%). L’expression des antigènes de surface CD4, CD56, TCL1, HLA-DR, TCF4 et CD123, de la chaîne α du récepteur de l’interleukine3, sur les cellules blastiques à l’immunohistochimie est caractéristique du BPDCN. La coexpression de CD123 et de TCF4 sur les cellules est typique. Toutefois, les marqueurs de surface spécifiques des lymphocytes B (p.ex. CD19), des lymphocytes T (p.ex. CD3) et des cellules myéloïdes (p.ex. MPO, CD14) ne doivent pas être détectables. L’expression anormale des marqueurs est également observée à la cytométrie en flux (Tab. 2).
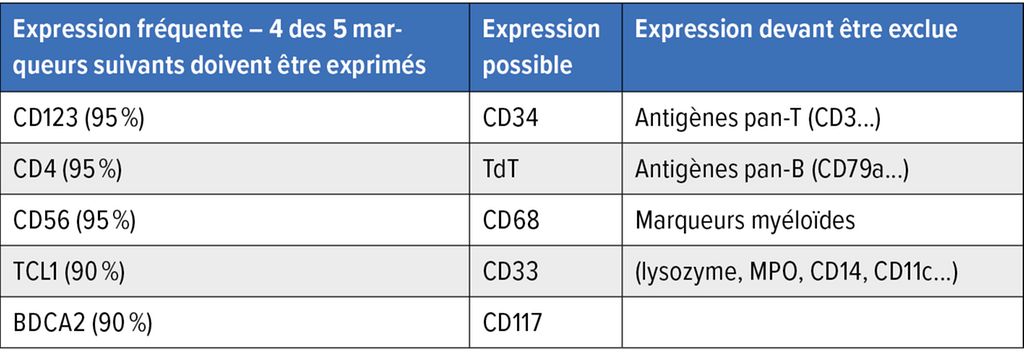
Tab. 2: Critères diagnostiques d’immunohistologie/de cytométrie en flux
Dans le cadre des examens initiaux, il est important de procéder à un diagnostic du liquide céphalo-rachidien, car environ 10% des patient·es présentent une atteinte du SNC au moment du diagnostic. Au cours de la maladie, une infiltration dans le liquide céphalo-rachidien survient chez jusqu’à 33% des personnes atteintes, bien que les symptômes typiques soient souvent absents.
De nombreux·ses patient·es présentent également un caryotype complexe ou des anomalies chromosomiques affectant 5q (70%), 12p (62%), 13q (60%) ou 6q (52%). Le séquençage nouvelle génération révèle souvent des mutations de TET2 (21–86%), d’ASXL1 (28–50%), de ZRSR2 (14–57%), de TP53 (38%), d’EZH2 (14%), de NRAS (14%), de NOTCH1 (14%), de SF3B1 (14%) et de SRSF2 (14%). Sur le plan du diagnostic différentiel, il est essentiel de distinguer le BPDCN de la LAM, et vice-versa. Les deux peuvent présenter des manifestations similaires en termes de tableau clinique et de morphologie, surtout en présence d’infiltrats cutanés dans la LAM.
Traitement
Compte tenu de l’évolution du BPDCN, la chimiothérapie a été utilisée en premier lieu jusqu’à présent. Il s’agissait le plus souvent de chimiothérapie intensive, comme dans le cas de la LAM ou de la leucémie aiguë lymphoblastique (LAL), ou de chimiothérapie standard, comme dans le cas des lymphomes non hodgkiniens (LNH) (Tab. 3). Tous les protocoles ont permis d’obtenir des rémissions chez une partie des patient·es. Compte tenu de la rareté de la maladie, aucune étude randomisée n’a été publiée à ce jour. Notre évaluation de l’efficacité des différents schémas thérapeutiques se fonde plutôt sur des données rétrospectives. En général, les traitements d’induction intensifs ont permis d’atteindre des taux de rémission plus élevés par rapport aux traitements standards (protocoles LNH). De plus, les protocoles LAL semblent être supérieurs aux protocoles LAM en termes de taux de réponse.
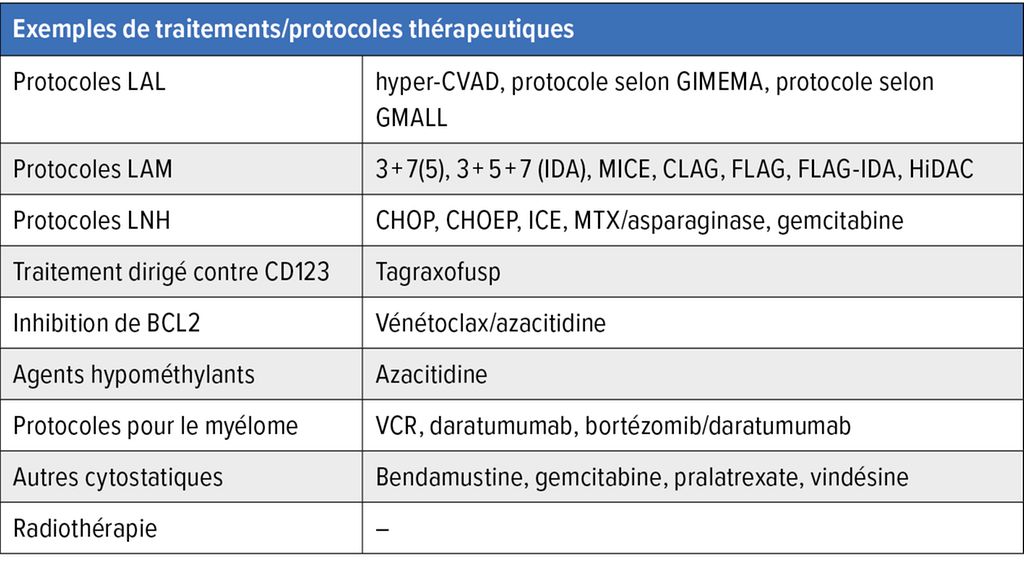
Tab. 3: Exemples de traitements/protocoles thérapeutiques utilisés auparavant et de nos jours en cas de BPDCN
Malgré une réponse modérée à bonne, un taux de récidive élevé conditionne une survie à long terme contrôlable, comme l’ont montré Laribe K et al. dans une analyse rétrospective des données de 398 patient·es issu·es de 75 centres, en 2022. La survie des patient·es a été de 18 mois avec les protocoles LAL/LAM, soit à peine plus longue qu’avec les protocoles LNH (14mois).
La seule option thérapeutique qui entraîne une prolongation significative de la survie est la greffe de cellules souches. Cela a pu être démontré pour la greffe allogénique comme autologue de cellules souches, mais la greffe autologue est ici inférieure à celle allogénique. Si possible, la greffe de cellules souches est donc le traitement de référence, l’obtention préalable d’une rémission complète étant essentielle pour la survie à long terme.
Le tagraxofusp a récemment été approuvé pour le traitement du BPDCN. Il s’agit d’une protéine de fusion associant l’interleukine-3 recombinante à une toxine diphtérique tronquée, qui utilise l’expression de CD123 sur les cellules du BPDCN. L’efficacité thérapeutique a pu être démontrée en 2019 dans le cadre d’une étude ouverte. Sur 29 patient·es recevant 12μg de tagraxofusp/kg de poids corporel après détermination de la dose, 72% ont présenté une réponse complète ou «clinique complète». Dans les données à long terme de 65 patient·es publiées en 2022, la réponse complète ou «clinique complète» était de 57% et la réponse globale (y compris la réponse partielle) de 75%. Chez 45% d’entre eux·elles, une greffe de cellules souches a pu être réalisée après l’obtention d’une rémission. Il convient de mentionner qu’un score ECOG<2 et une fonction organique adéquate étaient des conditions préalables à l’inclusion dans l’étude. Les effets secondaires suivants sont survenus, en particulier au cours du premier cycle de traitement: augmentation des transaminases, hypoalbuminémie, œdème périphérique, thrombocytopénie et le syndrome de fuite capillaire. Dans ce dernier cas, on a observé une baisse de l’albumine le premier jour de traitement. En cas de baisse de l’albumine, les directives recommandent d’interrompre la prise de tagraxofusp et de substituer l’albumine.
Un certain nombre d’autres nouveaux traitements ont également été utilisés avec succès dans le BPDCN, bien que seuls des séries de cas ou des rapports de cas n’aient été publiés à ce jour. Les exemples les plus connus sont l’association vénétoclax/azacitidine, l’azacitidine en monothérapie ou l’association bortézomib/daratumumab.
Chez les patient·es présentant une atteinte du SNC, un traitement par voie intrathécale approprié est nécessaire, comme en cas de LAL.
Résumé
Le BPDCN est une maladie rare. Le diagnostic doit être posé de manière multidisciplinaire par des dermatologues, des pathologistes et des hématologues. Il est essentiel de penser à cette maladie en présence de lésions cutanées typiques et d’initier la procédure diagnostique correspondante. En plus de la chimiothérapie conventionnelle, une nouvelle option thérapeutique est disponible avec le tagraxofusp pour permettre aux patient·es d’obtenir une rémission. Encore aujourd’hui, seule une greffe de cellules souches permet d’envisager une survie à long terme.
Littérature:
auprès des auteur·es
Das könnte Sie auch interessieren:
Le symposium «ASCO Genitourinary Cancers Symposium» fête son 20e anniversaire
Avec 5200inscriptions et 877résumés soumis, le symposium «ASCO Genitourinary Cancers Symposium» (ASCO GU) a fêté son 20eanniversaire au début de cette année. DrEric Small, président lors ...
La sélection est essentielle dans le traitement des tumeurs gastro-intestinales
La différenciation moléculaire au sein des entités tumorales a conduit à de nouvelles options thérapeutiques, en particulier en ce qui concerne les stratégies thérapeutiques ciblées. L’ ...
Nouveaux traitements de référence, thérapies combinées et séquençage optimal
L’European Lung Cancer Congress (ELCC) s’est tenu à Prague du 20 au 23 mars. Cette année encore, l’accent a été principalement mis sur les immunothérapies et les thérapies ciblées. Ci- ...